Cours de chimie Organique.
Eléments d'analyse conformationnelle
Définitions
Les stéréoisomères peuvent être classées selon leur mode d'interconversion :
- la configuration d'une molécule est la disposition de ses atomes dans l'espace indépendamment des rotations autour des liaisons simples ;
- les conformations d'une molécule sont les arrangements des atomes qui ne se différencient que par des rotations autour de liaisons simples. Ce terme a été introduit dans les années 30 par N. Haworth lors de son étude des sucres. L'étude systématique de la conformation des cycles de la famille du cyclohexane a été entreprise par le chimiste norvégien Odd Hassel. La prise en compte de l'analyse conformationnelle dans la prévision de la réactivité des stéroïdes a été reconnue dès les années 50 par le chimiste britannique Sir Derek H. R. Barton [1]. En 1969, le prix Nobel de chimie a été décerné à Hassel et Barton pour l'ensemble de leurs travaux sur ce sujet.
- les énantiomères ;
- les diastéréo-isomères.
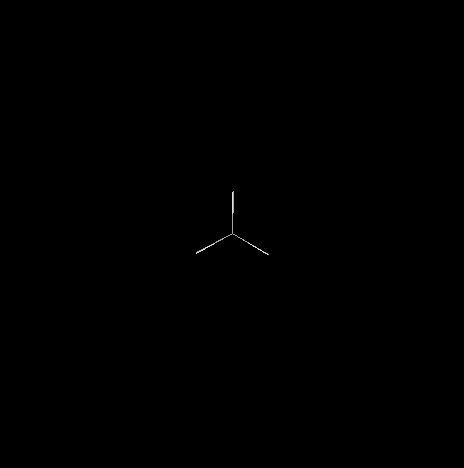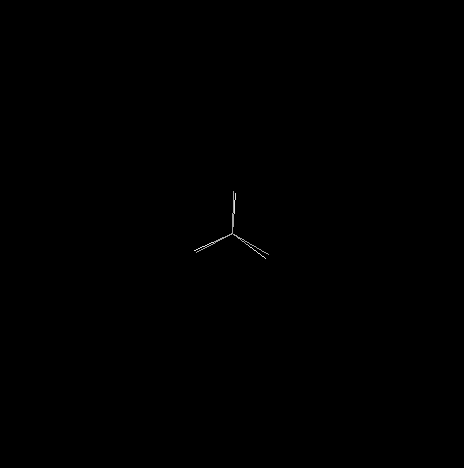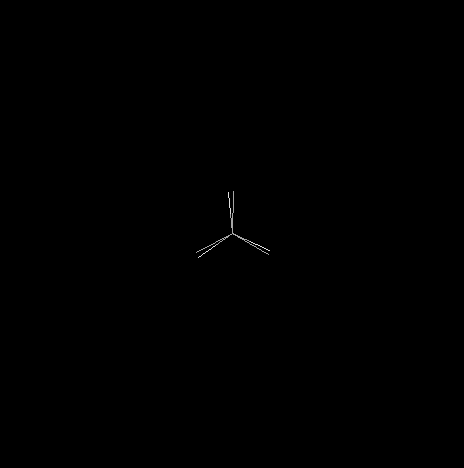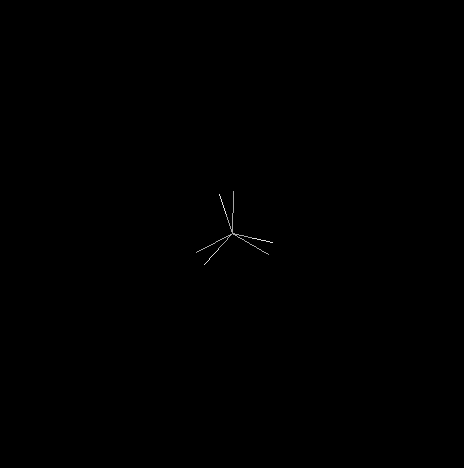
L'éthane est le deuxième terme de la série des alcanes. Sa molécule a pour formule brute C2H6.
 |
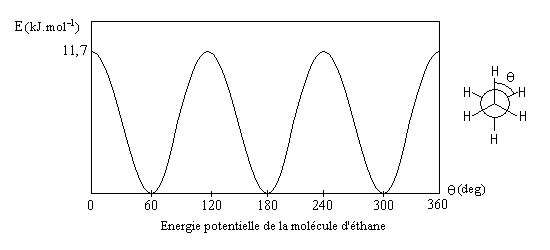
Le modèle représente la molécule d'éthane dans une conformation décalée. En observant la molécule en mode "spacefill", on constate que les atomes d'hydrogène ont un rayon de Van der Waals (120 pm) trop faible pour expliquer la différence d'énergie entre la conformations éclipsée et la conformation éclipsée. |
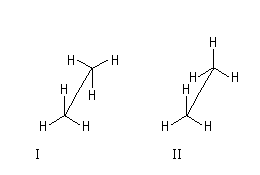
|
q (deg)
|
0
|
60
|
|
Conformation
|
II
|
I
|
|
Stéréodescripteur
|
synpériplanaire (éclipsée)
|
antipériplanaire (décalée)
|

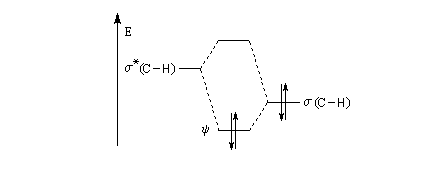
- la plus haute orbitale occupée est s(C-H) ;
- la plus basse orbitale vacante est s*(C-H).
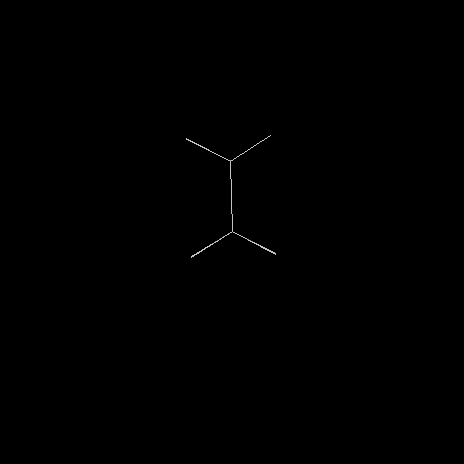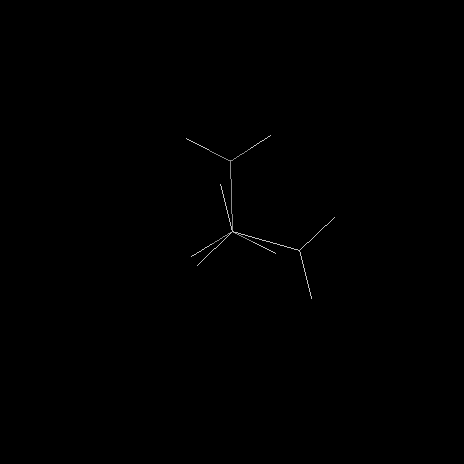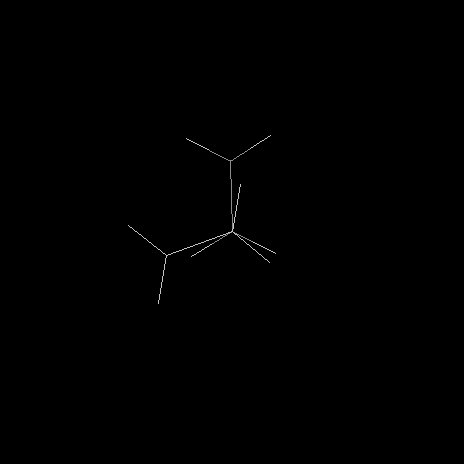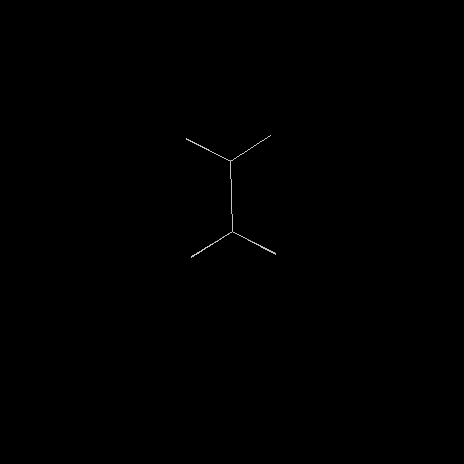
Le butane, dont la formule brute est C4H10 est le troisième terme de la série des alcanes.
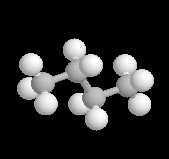 |
Le modèle de gauche représente la molécule de butane dans sa conformation antipériplanaire, correspondant à l'énergie la plus basse de la molécule. |
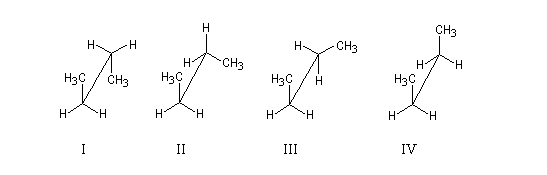
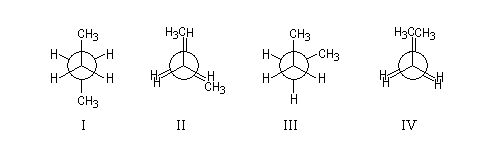
|
q (deg)
|
0
|
60
|
120
|
180
|
|
Conformation
|
IV
|
III
|
II
|
I
|
|
Nom
|
synpériplanaire
|
synclinale
|
anticlinale
|
antipériplanaire
|
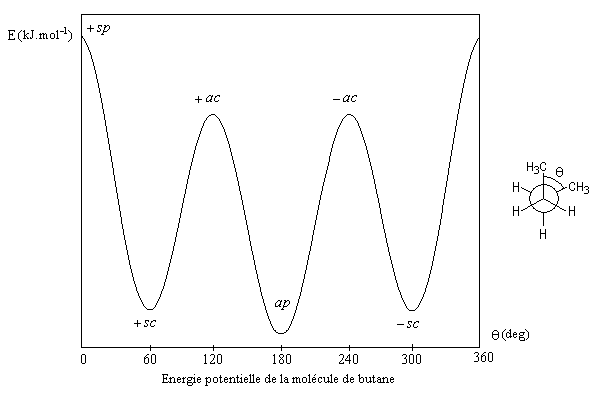
|
q (deg)
|
0
|
60
|
120
|
180
|
|
Energie (kJ.mol-1)
|
18,8
|
3,8
|
15,9
|
0
|
- la différence d'énergie entre la conformation antipériplanaire et la conformation anticlinale est essentiellement due à l'énergie de torsion, sa valeur est donc comparable à celle rencontrée pour l'éthane ;
- la différence d'énergie entre la conformation antipériplanaire et la conformation synpériplanaire est plus importante car à l'énergie de torsion s'ajoute l'énergie de répulsion entre les deux groupes méthyle dont les rayons de Van der Waals de 200 pm sont très supérieurs à celui de l'atome d'hydrogène ;
- il est utile de retenir la différence d'énergie de 3,8 kJ.mol-1 entre les conformations antipériplanaire et synclinale. On l'appelle aussi interaction butane gauche. On retrouve cette interaction dans de nombreuses structures organiques.
On peut reconnaître les enchaînements carbonés des molécules précédentes dans de nombreuses structures organiques. Le polyéthylène est obtenu par polymérisation de l'éthène (éthylène). Ses chaînes sont constituées de la succession de milliers de motifs -CH2-CH2-. Il existe deux grandes méthodes de polymérisation de l'éthène :
- La polymérisation ordinaire de l'éthène s'effectue par
voie radicalaire sous haute pression (2000 bar). Elle fournit des
macromolécules ramifiées car les radicaux intermédiaires arrachent des
atomes d'hydrogène de la chaîne donnant à leur tour d'autres radicaux.
Le polymère obtenu possède une
faible densité. En effet les chaînes ne peuvent pas s'approcher facilement les unes des autres.
- On peut obtenir un polyéthylène possèdant des chaînes linéaires en polymérisant l'éthylène à basse pression en présence de catalyseurs mis au point par Ziegler et Natta (prix Nobel de chimie 1963).
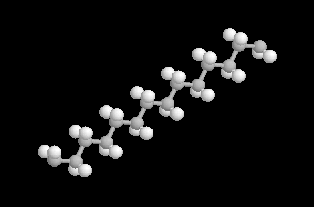 |
L'image de gauche représente un fragment de chaîne de polyéthylène linéaire. La macromolécule est constituée d'un enchaînement régulier d'atomes de carbone dont la conformation la plus stable est plane et en zigzag. Cette conformation permet aux chaînes de s'approcher très près les unes des autres ce qui explique la densité élevée du polymère si on la compare à celle du polyéthylène ramifié ordinaire. |
Diènes et polyènes
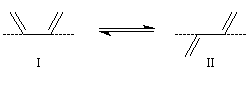
Les conformations dans lesquelles toutes les liaisons sont dans un même plan, sont privilégiées. Elles permettent un recouvrement maximal des orbitales p perpendiculaires au plan du système conjugué. Les stéréodescripteurs s-cis et s-trans se réfèrent à la position des liaisons doubles par rapport à la liaison simple s.
|
q (deg)
|
0
|
180
|
|
Conformation
|
I
|
II
|
|
Stéréodescripteur
|
s-cis
|
s-trans
|
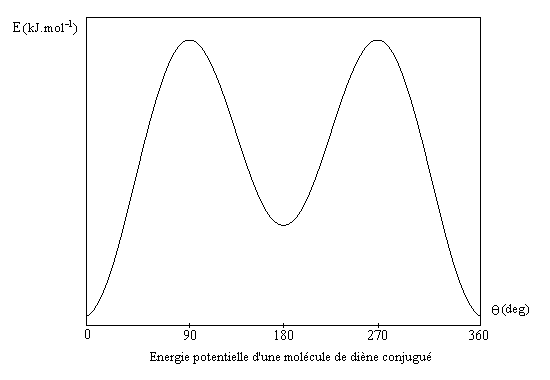
|
q (deg)
|
90
|
180
|
|
Energie (kJ.mol-1)
|
20,5
|
10,9
|
La conformation la plus stable est la s-trans.
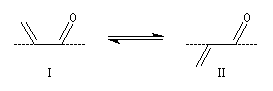
|
q (deg)
|
90
|
180
|
|
Energie (kJ.mol-1)
|
29,3
|
8,6
|
Cyclohexane
La formule brute du cyclohexane est C6H12. La molécule qui appartient à la famille des cyclanes possède une insaturation. La mesure de la chaleur de combustion indique que la tension dans le cycle est très faible. Comme l'a prédit Sachse dès 1860, la molécule ne doit pas avoir la forme d'un hexagone régulier sinon les angles entre deux liaisons adjacentes seraient 120° et on observerait une contrainte dans le cycle.
Des expériences de diffraction électronique en phase gazeuse ont confirmé que la conformation de plus basse énergie n'est pas plane. On l'appelle : conformation chaise.
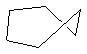
Parmi l'infinité des conformations possibles, celles dont la géométrie est remarquable sont classées ci-dessous par énergie croissante :
 |
 |
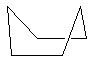 |
 |
|
chaise
|
bateau croisé
|
bateau
|
Enveloppe
|
La différence d'énergie entre ces la conformation chaise et la conformation bateau a deux origines :
- On reconnaît dans la conformation bateau des enchaînements de type butane synpériplanaire qui n'existent pas dans la conformation chaise où ces enchaînements sont de type butane synclinal.
- Il existe une répulsion importante entre les atomes d'hydrogène situés sur les mâts de la conformation bateau.
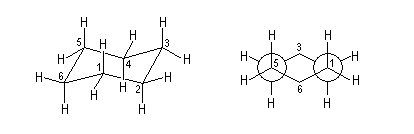
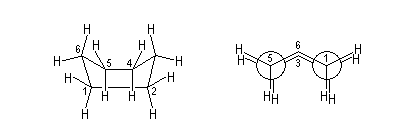
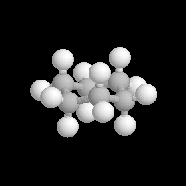 |
Dans cette conformation relativement rigide, les liaisons peuvent être classées en deux catégories :
|
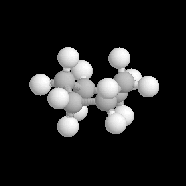 |
Dans la conformation bateau, relativement flexible, on distingue quatre catégories de liaisons :
|
La constante de couplage 3JHH entre deux protons vicinaux dépend de la valeur de l'angle dièdre entre les liaisons C-H. Cette dépendance peut être exprimée quantitativement grâce à la formule de Karplus.
Basculement conformationnel
Le passage d'une conformation chaise à une autre, inverse les positions axiales et équatoriales. A 300 K le passage d'une forme à l'autre s'effectue environ 100 000 fois par seconde.
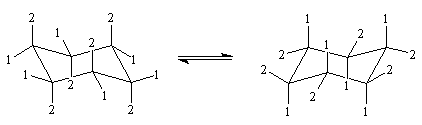
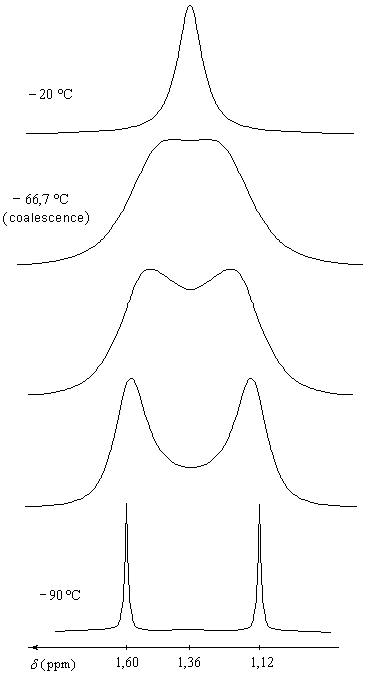 |
L'allure du spectre RMN du cyclohexane dépend de la température de travail :
|
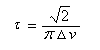
Les dérivés monosubstitués du cyclohexane ont été particulièrement étudiés par D. H. R. Barton et O. Hassel qui ont obtenu le prix Nobel de chimie en 1969. Désignons le substituant par X. Il existe deux conformations chaise en équilibre :
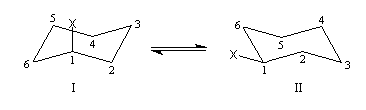
- Dans (II), la liaison C-X et les liaisons C2-C3 et C5-C6 forment un enchaînement de type butane antipériplanaire. En revanche, dans (I), la liaison C-X et les liaisons C2-C3 et C5-C6 forment un enchaînement de type butane synclinal.
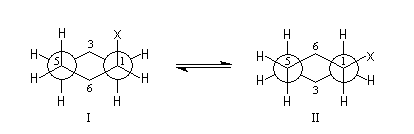
- Il existe des répulsions entre le groupe X et les atomes d'hydrogène situés en position axiale (interaction 1, 3 diaxiales) dans (I) qui n'existent pas dans (II). Leur intensité dépend de l'encombrement stérique du groupe X.
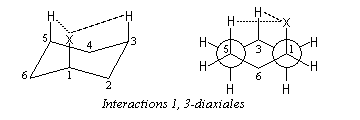
A = R.T ln K
On peut déterminer expérimentalement la valeur de A en utilisant différentes techniques, notamment la RMN. Soit P une propriété physique du système en équilibre, par exemple le déplacement chimique
du proton porté par l'atome de carbone 1. Supposons que
la vitesse d'interconversion soit beaucoup plus rapide que la vitesse
de mesure du spectromètre. Celui-ci enregistre alors une grandeur
moyenne telle que :|
Groupe X
|
méthyle
|
isopropyle
|
hydroxyle
|
tertiobutyle
|
|
A (kJ.mol-1)
|
7,5
|
9
|
3,3 (AcOH 75 %)
|
23,5
|
La valeur de A très élevée pour le groupe tertiobutyle montre que dans le cas du tertiobutylcyclohexane seule la conformation plaçant ce groupe en équatoriale est possible.
 |
Le groupe tertiobutyle est si volumineux qu'il ne peut exister qu'en position équatoriale.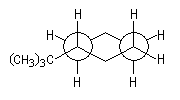 Modèle interactif du tertiobutylcyclohexane. |
Lorsque le cycle est disubstitué, on peut considérer en première approximation que les valeurs de A relatives à chaque substituant sont additives. On peut ainsi déterminer par le calcul la constante d'équilibre de l'interconversion entre les cycles disubstitués.
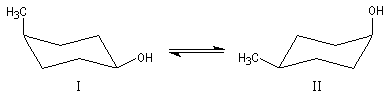
DrGo = - 4,2 kJ.mol-1
K = [II]/[I]
cela montre que les molécules qui adoptent la conformation II sont en plus grand nombre que celles qui adoptent la conformation I.
Comme dans le cas du cyclohexane, la vitesse du basculement conformationnel diminue quand la température diminue. On donne ci-dessous quelques valeurs des temps de demi-réaction t pour l'inversion de conformation du chlorocyclohexane.
|
Température (°C)
|
Temps de demi-réaction t
|
|
25
|
1,3 10-5 s
|
|
-120
|
23 min
|
|
-160
|
22 ans
|
Décalines
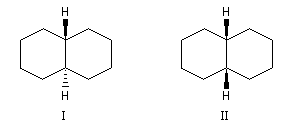
Les décalines sont des hydrocarbures de formule brute C10H18. Ces molécules sont constituées de deux cycles cyclohexaniques accolés. On peut donc les considérer comme des dérivés disubstitués particuliers du cyclohexane.
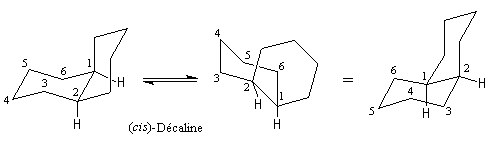
A partir de l'un des cycles, les liaisons constituant l'amorce du deuxième cycle peuvent être de stéréochimie trans (I) ou bien de stéréochimie cis (II). Le passage de (I) à (II) nécessite la rupture et la reformation de liaisons. Ce sont des isomères de configuration et plus précisément des diastéréo-isomères.
Le cas rappelle celui des diastéréo-isomères cis et trans du 1, 2-diméthylcyclohexane qu'il est conseillé d'étudier avant d'aborder ce paragraphe.
|
Composé
|
(trans)-décaline (I)
|
(cis)-décaline (II)
|
|
TF (°C)
|
- 32
|
- 42
|
|
Indice de réfraction (raie D, Na)
|
1,4690
|
1,480
|
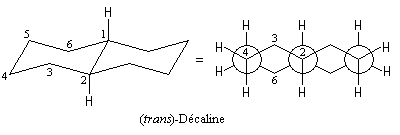
 |
La décaline trans possède un centre de symétrie au milieu de la liaison C1-C2. Il s'agit donc d'une molécule achirale. |
 |
La décaline cis, considérée comme une structure figée est chirale. Cependant, le basculement conformationnel fait passer d'une conformation à son image dans un miroir. L'énergie séparant les deux conformations énantiomères est d'environ 25 kJ.mol-1. A la température ordinaire le passage d'une conformation à l'autre est très aisé et l'on observe le mélange racémique des deux énantiomères. |
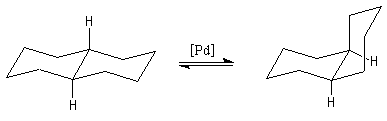
DrHo = 11,3 kJ.mol-1
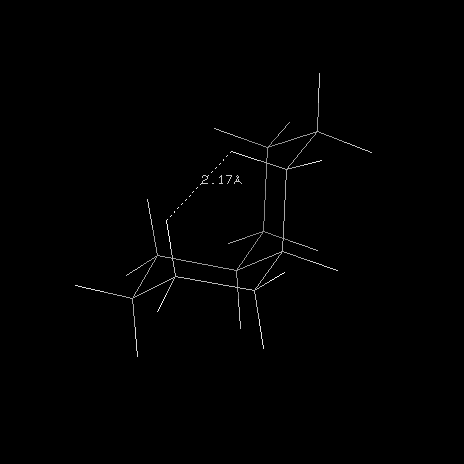
L'une des causes principales de l'instabilité de la cis décaline par rapport à son isomère trans
vient du fait que les atomes d'hydrogène portés par la face concave
sont à une distance de 0,217 nm qui est inférieure à la somme de leurs
rayons de Van der Waals 0,240 nm.Stéroïdes
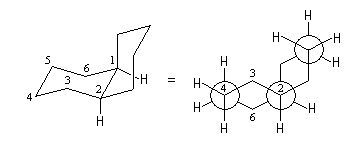
Les stéroïdes sont des composés comportant un squelette carboné constitué d'un système de type phénantrène (cycles A, B, C) avec un cycle cyclopentane accolé (cycle D).
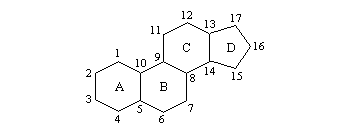
Par déshydratation suivie d'une hydrogénation du cholestérol, on peut obtenir deux hydrocarbures diastéréo-isomères représentés ci-dessous. A l'image de ce qui se passe pour les décalines, la jonction entre les cycles A et B est de type trans dans le premier et cis dans le second.

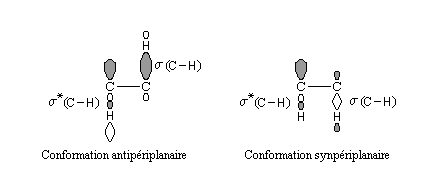
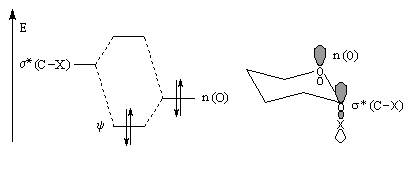
Lorsqu'on étudie l'équilibre conformationnel des tétrahydropyrannes substitués en position 2 par un groupe électronégatif, on constate que la conformation dans laquelle X est en position axiale est majoritaire à l'équilibre. Ce phénomène porte le nom d'effet anomère.
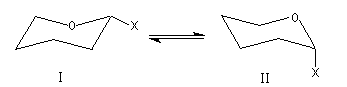
l'interaction entre les orbitales est maximale pour une disposition axiale de l'orbitale décrivant le doublet non liant et de l'orbitale antiliante s*(C-X). Il s'agit d'un exemple d'hyperconjugaison.
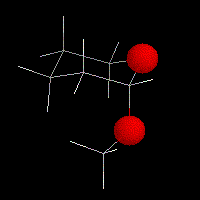 |
Chez les 2-alcoxy tétrahydropyrannes comme celui qui est représenté à gauche, l'effet anomère se traduit par la disposition axiale adoptée par le groupe alcoxy situé en position 2. L'effet est très répandu dans la chimie des sucres. |
 |
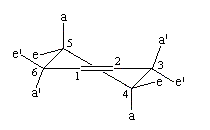 |
- axiaux a ;
- pseudo-axiaux a' ;
- équatoriaux e ;
- pseudo-équaoriaux e'.
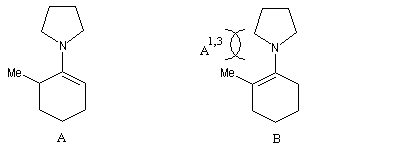
Les énamines sont des intermédiaires de synthèse utilisés dans l'alkylation régiosélective des cétones. Tension allylique
La tension allylique (en anglais : allylic strain) résulte d'une interaction au sein d'une molécule entre substituants d'une double liaison. L'un de ces substituants étant en position allylique. A l'origine le phénomène a été décrit chez les dérivés substitués du cyclohexène. On distingue deux types appelés respectivement tension allylique A1,2 et tension A1,3 (selon les notations de F Johnson et S. K. Malhotra qui ont décrit le phénomène.)
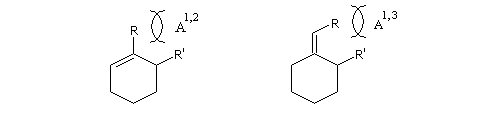
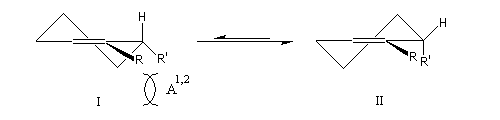
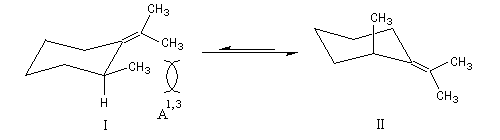
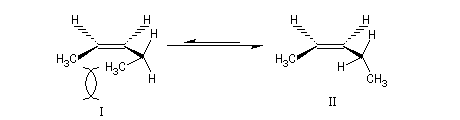
L'existence du pont permet d'isoler des molécules dans lesquelles un cycle à 6 chaînons adopte les géométries variées rencontrées chez le cyclohexane.
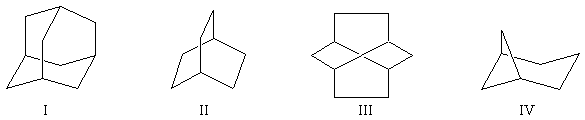
|
molécule
|
I
|
II
|
III
|
IV
|
|
nom
|
bicyclo [2, 2, 2]-octane
|
twistane
|
bicyclo [3, 1, 1]-heptane
|
|
|
cycle
|
chaise
|
bateau
|
twist
|
chaise et bateau
|
Cette règle d'origine expérimentale affirme l'impossibilité d'existence d'une double liaison en tête de pont d'un composé bicyclique. On en retrouve la manifestation chez certaines cétones bicycliques dont le taux d'énolisation est nul. Lorsque les cycles sont assez grands il est possible de préparer des composés comportant une double liaison en tête de pont. Le premier composé de ce type a été préparé grâce à la réaction d'aldolisation par Prelog en 1950.
Si vous disposez de Firefox ou I.E ver > 6.0 associés au plug-in Chime 2.6 SP4 (MDL) vous pouvez compléter l'étude du cyclohexane et de ses dérivés en ouvrant la page suivante : dérivés monosubstitués du cyclohexane. Bibliographie
Ouvrages théoriques
J. March - Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R.J. Sundberg - Advanced organic chemistry, 3d edition (Plenum Press, 1990).
H. Kagan - La stéréochimie organique (PUF, 1975).
J. L Pierre - Principes de stéréochimie organique statique (A. Colin, 1971).
D. H. R. Barton, Some recollections of gap jumping (American Chemical SOciety, 1991).
A. Kirmann, J. Cantacuzène, P. Duhamel - Chimie organique T 1 (A. Colin, 1971).
K. Mislow - Introduction to stereochemistry (W. A Benjamin, New York, 1966).
J. Jacques - La molécule et son double (Hachette, 1992).
V. Pellegrin - Les représentations graphiques bidimensionnelles des molécules en chimie organique (Bulletin de l'Union des Physiciens, février 1999).
Sur la toile
Cours de stéréochimie de J. C. Gressier
Termes utilisés en stéréochimie
The principles of conformationnal analysis (Sir D. Barton, The Nobel Prize in Chemistry 1969)
Sir N. Haworth, The Nobel Prize in Chemistry 1937
Derek Barton speaking on conformational analysis
A-values
Dynamic NMR----
Martin Karplus
Steric Effects Don't Explain Ethane Conformation
K. Ziegler and G. Natta the Nobel Prize in Chemistry 1963
Aspects of the determination of equilibration rates by NMR spectroscopy. by J. D. Roberts, California Institute of Technology, Pasadena, California, USA.
Articles
[1] The conformation of the steroid nucleus, by D.H.R. Barton, Cambridge, Mass.
[2] Pophristic, V., and L. Goodman. Hyperconjugation not steric repulsion leads to the staggered structure of ethane. Nature 411 2001, May 31, 565.
[3] F. R. Jensen, C. H. Bushweller, J. Am. Chem. Soc.91, 3223 (1969).
[4] H. S. Gutowsky and C. H. Holm, J. Chem. Phys. 25. 1228. (1956).
J. R. Wiseman, J. Am. Chem. Soc. 1967, 89, 5966. (exception Bredt)
J. A. Marshall, H. Faubl, J. Am. Chem. Soc. 1967, 89, 5965 .(exception Bredt)
Francis Johnson, Sudarshan K. Malhotra J. Am. Chem. Soc., 1965, 87 (23), pp 5492–5493
A synthetic attack on the oestrone problem, Woodward, R. B. (Robert Burns), Thesis (Ph. D.)--Massachusetts Institute of Technology, Dept. of Chemistry, 1937.









{kind=link}
{kind=link}
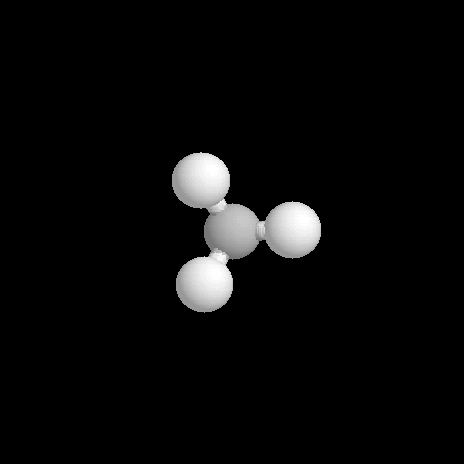{kind=link}
{kind=link}
{kind=link}
0 commentaires:
Enregistrer un commentaire