Cours de chimie Organique -
Aldéhydes & Cétones
Généralités
Définitions Les composés comportant dans leur molécule le groupe carbonyle -CO- sont appelés composés carbonylés. Il leur correspond deux fonctions :
- les aldéhydes comportent le groupe caractéristique -CHO. Le groupe carbonyle est lié à un groupe alkyle ou phényle et un atome d'hydrogène (exceptionnellement deux atomes d'hydrogène dans le cas du méthanal) ;
- les cétones comportent le groupe caractéristique -CO-R. Le groupe carbonyle est lié à des groupes alkyles ou phényles.
Etat naturel
 |
La civétone est une cétone à grand cycle présente à l'état naturel dans les glandes odoriférantes de la Civette ou chat musqué d'Afrique. Sa structure a été déterminée par le chimiste suisse d'origine croate L. S. Ruzicka en 1926 (prix Nobel 1939). Au cours de cette étude, Ruzicka mit au point une méthode de synthèse des cétones à grand cycle par chauffage de sels de thorium de diacides carboxyliques, une variante de la réaction de Piria. La synthèse totale de la civétone a été réalisée par M. Stoll en 1948. D'autres cétones à grands cycles sont utilisées en parfumerie comme la cyclopentadécanone (exaltone) ou musc artificiel |
 |
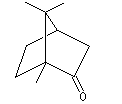
Le camphrier est un arbre appartenant à la famille des lauracées originaire du Japon et de Chine. Cet arbre, dont la hauteur peut atteindre plus de 40 m, a une longévité de plusieurs centaines d'années. Les feuilles du camphrier (image de gauche) rappellent celles du laurier. Le camphre naturel (Cinnamomum Camphora) est extrait de l'huile du bois de camphrier. Il est utilisé en médecine comme révulsif et tonicardiaque. La formule brute du camphre a été établie par J. B. Dumas en 1831. Sa synthèse a été effectuée par Komppa en 1903. L'étape clef est une condensation de Claisen |
 |
Le camphre naturel est le (-)(1R, 4R)-triméthyl-1,7,7-bicyclo [2, 2, 1] heptan-2-one. La molécule possède
deux atomes de carbone asymétriques.
 |
Carbonylés importants
 |
Le méthanal est préparé par oxydation du méthanol.
|
 |
L'éthanal préparé actuellement par oxydation de l'éthène grâce au procédé Wacker.
|
 |
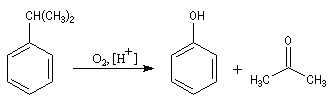
La propanone est préparée à partir du propène par le procédé Wacker. C'est aussi un sous-produit de la synthèse du phénol par le procédé Hock dont le bilan
revient à oxyder le cumène par l'oxygène de l'air.
 |
 |
La cyclohexanone est préparée par oxydation du cyclohexanol. Elle est utilisée comme
solvant. Son oxime permet la synthèse du caprolactame, matière première de la synthèse du Nylon 6 obtenu à la suite d'une transposition de Beckmann.
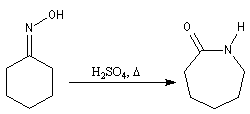 |
Nomenclature
Aldéhydes
On distingue les séries acyclique et cyclique.
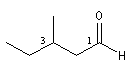
- Série acyclique
Le nom est formé en ajoutant le suffixe al au nom de l'hydrocarbure correspondant à la chaîne principale. L'atome de carbone du groupe porte le numéro 1 qui n'est pas mentionné.
 |
 |
|
hexanal
|
3-méthylpentanal
|
 |
 |
|
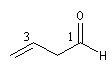
hexanedial
|
but-3-énal
|
- Série cyclique
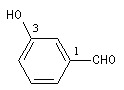
Lorsque le groupe CHO est fixé sur un atome de carbone faisant partie d'un système non aromatique on ajoute la terminaison carbaldéhyde au nom du système cyclique.
Lorsque le groupe CHO est fixé sur un cycle benzénique, le composé est nommé comme dérivé du benzaldéhyde.
|
|
 |
|
cyclohexanecarbaldéhyde
|
3-hydroxybenzaldéhyde
|
 |
 |
|
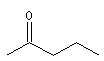
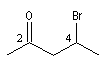
pentan-2-one
|
4-bromopentan-2-one
|
 |
 |
|
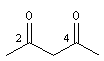
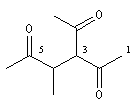
pentane-2, 4-dione
|
3-acétyl-4-méthylhexane-2, 5-dione
|
- Série cyclique
 |
 |
|
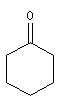
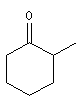
cyclohexanone
|
2-méthylcyclohexanone
|
 |
 |
|
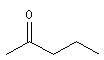
méthylpropylcétone
|
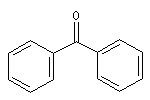
diphénylcétone
|
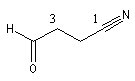 |
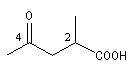 |
|
3-formylpropanenitrile
|
acide 2-méthyl-4-oxopentanoïque
|
Constantes physiques Voici quelques résultats expérimentaux concernant le moment dipolaire et les énergies de liaisons de quelques composés carbonylés.
|
Molécule
|
HCHO
|
CH3CHO
|
CH3COCH3
|
|
p (D)
|
2,27
|
2,73
|
2,84
|
|
Liaison
|
C-C
|
C-O
|
C=C
|
C=O
|
|
D (kJ.mol-1)
|
347
|
360
|
610
|
740
|
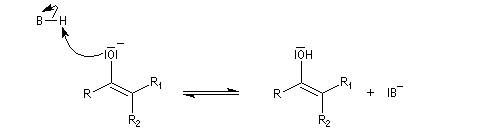
- on note une valeur élevée du moment dipolaire des molécules qui traduit la polarité importante du groupe carbonyle ;
- l'énergie de dissociation particulièrement élevée de la double liaison CO qui est supérieure au double de celle d'une liaison simple entre ces atomes.
|
Composé
|
TF (°C)
|
TE (°C)
|
d
|
|
HCHO
|
- 118
|
19
|
-
|
|
CH3CHO
|
- 123
|
21
|
0,79
|
|
CH3CH2CHO
|
- 81
|
48
|
0,80
|
|
CH3COCH3
|
- 95
|
56
|
0,79
|
Structure électronique du groupe carbonyle
Méthode des orbitales moléculaires Le groupe carbonyle peut être décrit par deux méthodes qui sont souvent utilisées conjointement et qui se complètent de façon utile. La méthode des orbitales moléculaires et celle, plus fruste, de la mésomérie.
Nous donnons ci-dessous un diagramme qualitatif des orbitales moléculaires pour une molécule de méthanal. Les orbitales moléculaires des autres composés carbonylés peuvent être obtenues par la méthode des perturbations.
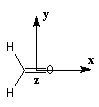 |
Les expériences de diffraction montrent que les
atomes de carbone, d'oxygène et d'hydrogène sont situés dans un même
plan. La molécule
présente deux plans de symétrie :
|
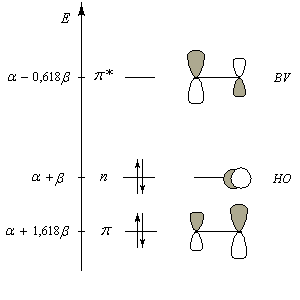 |
Le diagramme d'orbitales moléculaires représenté à gauche regroupe les OM p liante, n non liante et p* antiliante. Les orbitales frontières sont :
|
Rappelons que : a < 0 et b < 0.
|
aC
|
aO
|
bC-O
|
|
a
|
a + b
|
1,1 b
|
Avec ces valeurs les expressions des orbitales moléculaires p du méthanal s'écrivent :
|
Orbitale moléculaire
|
Energie
|
|
p : j1 = 0,85 pO + 0,53 pC
|
E1 = a + 1,618 b
|
|
p* : j2 = 0,53 pO - 0,85 pC
|
E2 = a - 0,618 b
|
Méthode de la mésomérie Dans la méthode de la mésomérie, le groupe carbonyle est décrit par deux formes limites principales représentées ci dessous. Celle de gauche est la forme ylène. Celle de droite présente une séparation des charges. L'atome d'oxygène, plus électronégatif que l'atome de carbone, supporte la charge négative. Cette forme est appelée forme ylure (en anglais : ylid).
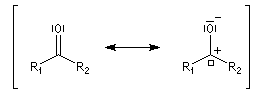
Spectroscopie
Spectroscopie infrarouge
C'est une méthode de choix pour l'étude des composés carbonylés. Le nombre d'onde de la vibration de valence du groupe carbonyle dépend du type de composé mais apparaît nettement sous la forme d'une bande intense dans une région assez dégagée du spectre.
L'exemple ci-dessous concerne la 4-méthylpentan-2-one.
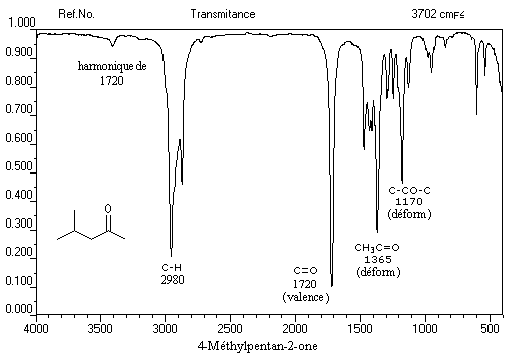
|
s (cm-1)
|
2700 - 2900
|
1650 - 1730
|
|
vibration
|
élongation C - H
|
élongation C = O
|
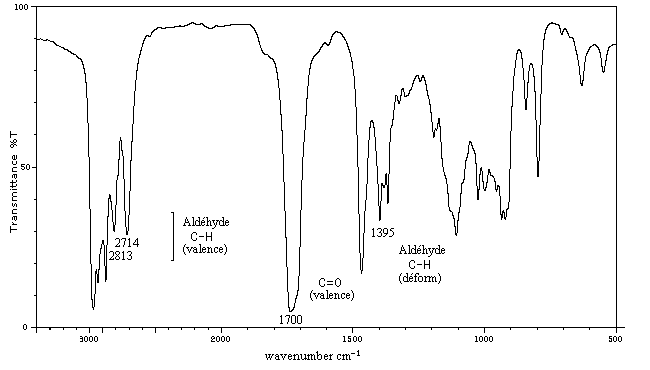
|
Composé
|
Cyclohexanone
|
Cyclopentanone
|
Cyclobutanone
|
|
s (cm-1)
|
1685-1705
|
1740-1750
|
1780
|
L'effet inductif attracteur du groupe carbonyle diminue la densité électronique autour du noyau d'hydrogène. Voici quelques valeurs de déplacement chimique :
|
Protons
|
H3C-CO-
|
-CH2-CO-
|
H-CO-
|
|
d (ppm)
|
2
|
2,5
|
10
|

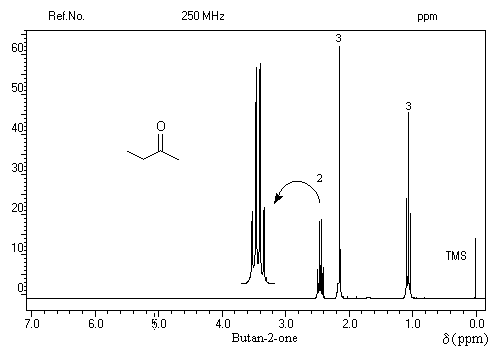
- ils sont liés au groupe carbonyle très électronégatif ;
- l'anisotropie du champ magnétique due au courants de cycle.

- les trois protons du méthyle sortent sous la forme d'un doublet à d = 2,2 ppm ;
- le proton aldéhydique se manifeste par un quadruplet à d = 9,8 ppm.

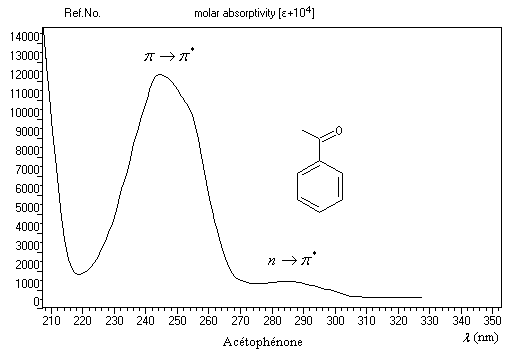
Les transitions utilisables avec les appareils courants sont celles qui impliquent les niveaux p, n et p*.
Transition p ® p*
|
Composé
|
lm (nm)
|
log e
|
Solvant
|
|
CH3CHO
|
193
|
-
|
cyclohexane
|
|
CH3COCH3
|
188
|
3,04
|
heptane
|
|
Composé
|
lm (nm)
|
log e
|
Solvant
|
|
CH3CHO
|
290
|
1,23
|
cyclohexane
|
|
CH3COCH3
|
279
|
1,17
|
heptane
|
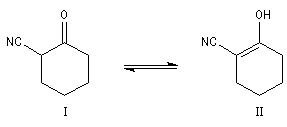
|
Composé
|
I
|
II
|
|
lm (nm)
|
290
|
230
|
|
Pourcentage
|
75
|
25
|
- le niveau p est peu affecté ;
- le niveau excité p*, plus polaire est abaissé ;
- le niveau n subit un abaissement d'énergie assez important.
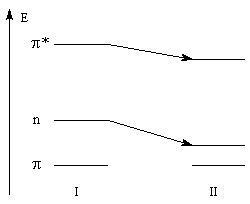
- la longueur d'onde du maximum d'absorption de la transition p ® p* est déplacée vers les grandes longueurs d'onde (effet bathochrome) ;
- la longueur d'onde du maximum d'absorption de la transition n ® p* est déplacée vers les faibles longueurs d'onde (effet hypsochrome).
Activation électrophile
Lorsqu'on dissout de la benzophénone dans l'acide sulfurique concentré, la solution obtenue a une couleur rouge. On interprète cette apparition de la couleur par l'existence dans l'acide sulfurique d'une forme protonée qui accroit les possibilités de délocalisation électronique.
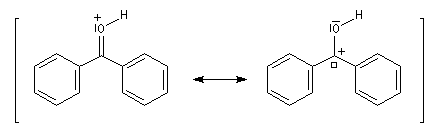
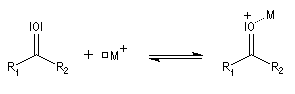
- Méthode de la mésomérie.
L'association entre l'oxygène et M+ augmente le poids relatif de la forme mésomère possédant un atome de carbone chargé positivement car l'existence de cette forme n'est plus subordonnée à une séparation de charge.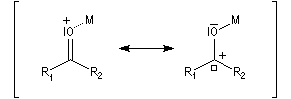
- Méthode des orbitales moléculaires.
Le diagramme d'orbitales moléculaires, montre que la plus basse orbitale vacante est l'orbitale moléculaire p*.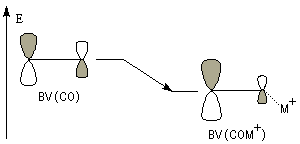
Le phénomène que nous venons de décrire, porte le nom d'assistance électrophile.
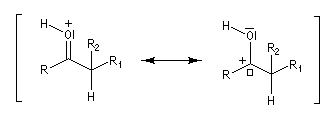
Géométrie de l'addition nucléophile Commençons par examiner la direction d'attaque du nucléophile dans le plan perpendiculaire au plan du carbonyle.
Avec les nucléophiles peu encombrés, la géométrie de l'addition est gouvernée par le recouvrement des orbitales frontières des réactifs. Raisonnons dans le cas de l'addition d'un ion hydrure. Ce dernier intervient par sa plus haute om occupée (1 s). Le carbonyle intervient par la plus basse om vacante. Le schéma ci-dessous résume la situation en l'absence (I) puis en présence d'une activation électrophile (II).
Le phénomène a été étudié dans les années 70 par H. B. Bürgi, J. D. Dunitz et J. M. Lehn [43]. L'angle d'attaque, appelé usuellement angle de Bürgi-Dunitz, est voisin de 105° (figure I). Ce résultat s'interprète en considérant le recouvrement des orbitales. La géométrie de l'interaction maximise l'interaction stabilisante tandis qu'elle minimise l'interaction secondaire déstabilisante entre les lobes orbitalaires de signes contraires (cette interaction est figurée en pointillés sur la figure.)
En présence d'activation électrophile par l'acide de Lewis M+, le coefficient sur l'atome de carbone augmente ce qui a pour conséquence d'augmenter l'intensité de l'interaction prinicpale stabilisante et de diminuer l'interaction secondaire déstabilisante. L'angle d'attaque diminue et passe à une valeur voisine de 95° (figure II.)
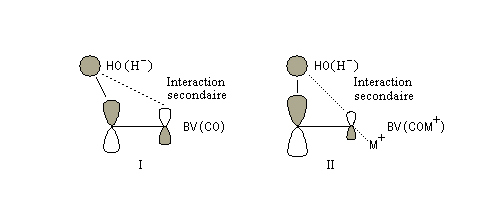
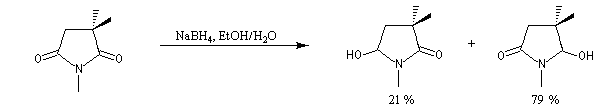
Remarque : l'angle entre la direction d'attaque du nucléophile et celui du carbonyle dans le plan joue également un rôle dans le cas où l'on a affaire à des aldéhydes possèdant un atome de carbone chiral en a du carbonyle (angle de Flippin-Lodge). Cet effet est étudié dans le chapitre consacré à la stéréochimie dynamique.
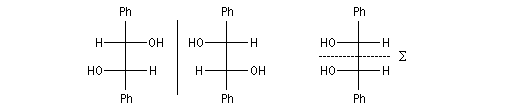
Cas où les faces du carbonyle sont prochirales
Les deux faces d'un groupe carbonyle lié à deux substituants différents sont prochirales et ces faces sont nommées en utilisant la convention de Hanson. L'exemple ci-dessous concerne la molécule d'éthanal.
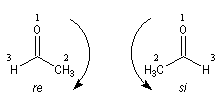
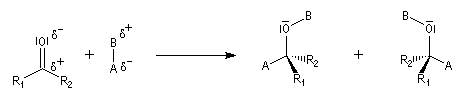
- les faces sont énantiotopiques comme, par exemple, dans la molécule d'éthanal, on obtient deux énantiomères ;
- les faces sont diastéréotopiques c'est le cas si R1 ou R2 sont chiraux, on obtient deux diastéréo-isomères.
Présence d'un carbone asymétrique en a du groupe carbonyle
La présence d'un centre chiral en a du groupe carbonyle pose un problème d'induction asymétrique, la règle de Cram ainsi que les modèles de Felkin et de Felkin-Anh sont examinés dans le chapitre consacré à la stéréochimie dynamique.
Addition nucléophile des hydrures Réactifs donneurs d'hydrure L'ion hydrure contient l'élément hydrogène au degré (-I). C'est donc un réducteur potentiel de fonctions insaturées. Les hydrures alcalins LiH, NaH, KH sont connus depuis longtemps. Cependant ces composés ne sont guère utilisables comme réducteurs car ce sont des bases très fortes et l'ion H- n'y manifeste pas de propriétés nucléophiles. En revanche, les complexes d'hydrures d'aluminium ou de bore, réactifs gros et polarisables, permettent de transférer l'ion H- vers un substrat insaturé en exaltant sa capacité nucléophile.
Le tétrahydruroaluminate de lithium LiAlH4 est un composé ionique qui se présente sous la forme d'un solide blanc, stable dans l'air sec mais extrêmement réactif. Il a été préparé en 1947 par H. J. Schlesinger. On l'utilise dans l'éthoxyéthane ou le THF. Il est absolument incompatible avec l'eau qu'il réduit avec explosion à cause du dégagement d'hydrogène et du caractère très exothermique de la réaction. On peut le préparer par la réaction suivante :
 |
Le tétrahydroborate de sodium NaBH4 (borohydrure de sodium) est un agent réducteur beaucoup plus doux que LiAlH4. Il a été introduit en synthèse organique par H. C. Brown. On peut le préparer en faisant réagir l'hydrure de sodium et le diborane. |
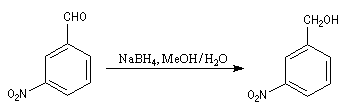
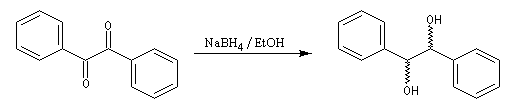
A ces réactions on peut rattacher la réaction de Meerwein, Ponndorf et Verley dans laquelle l'isopropylate d'aluminium permet le transfert d'un ion hydrure vers un composé carbonylé. Les réactions de réduction des carbonylés impliquant des organomagnésiens encombrés procèdent d'un mécanisme assez comparable.
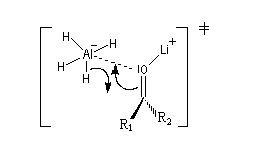
Bilan et mécanisme Le cation intervient dans l'activation électrophile du groupe carbonyle. Une preuve de cette activation est fournie par le fait expérimental suivant : lorsqu'on complexe le cation Li+ par un cryptand de taille convenable on observe un ralentissement très net de la réaction.
Un état de transition plausible pour le transfert de l'ion hydrure vers le groupe carbonyle est représenté ci-dessous :
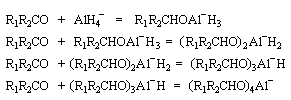
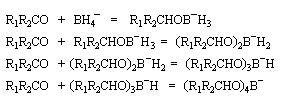
- en l'absence d'interaction d'acide-base de Lewis, on utilise le modèle de modèle de Felkin-Anh (1977) ;
- dans le cas contraire, il faut envisager un contrôle de la stéréochimie par chélation.
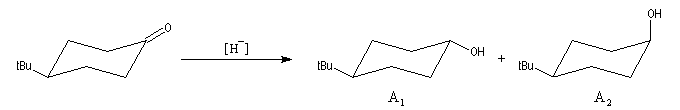
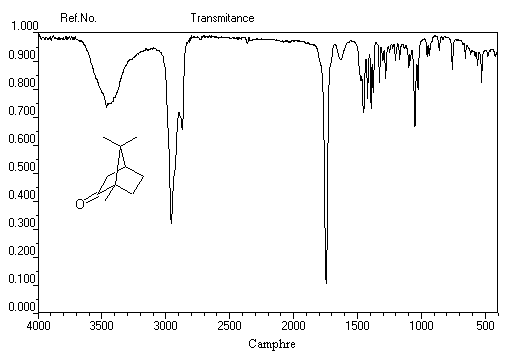
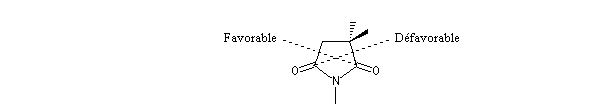
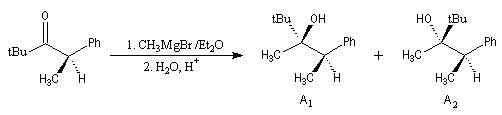
Le cas des composés bicycliques pontés est un plus délicat. Expérimentalement, on constate que les facteurs stériques sont prédominants et l'hydrure est transféré de façon préférentielle sur la face moins encombrée. L'exemple suivant concerne la réduction de la molécule de camphre qui donne un mélange dans lequel l'isobornéol (composé A1) pour lequel le groupe OH est situé sur la même face que le pont ( face exo) est largement majoritaire sur le bornéol (composé A2) pour lequel le groupe OH est situé sur la face opposée au pont (face endo).
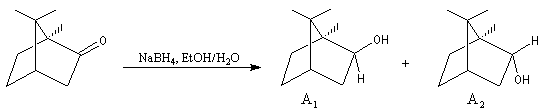
|
Composé
|
A1
|
A2
|
|
NaBH4
|
86
|
14
|
 |
Le modèle ci-contre représente un borohydrure alkylé commercialisé sous le
nom de K-Sélectride (Aldrich). On l'obtient en faisant réagir un trialkylborane sur l'hydrure
de potassium. |
Résultats expérimentaux Le produit d'addition de l'eau sur un composé carbonylé est un gem-diol.
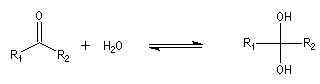
K = [hydrate]/[carbonylé]
|
Composé
|
CH3COCH3
|
CH3CHO
|
HCHO
|
F3CCHO
|
|
K
|
1,4´10-3
|
1,06
|
2,3´103
|
2,9´104
|
- l'atome de carbone d'un aldéhyde est moins encombré que celui d'une cétone ;
- cet atome est moins substitué par des groupes donneurs par effet inductif. Il est plus déficient en électrons et donc plus électrophile ;
- notons la valeur élevée de la constante thermodynamique K pour le méthanal qui se distingue des autres aldéhydes par une plus grande réactivité.
- catalyse acide ;
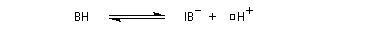
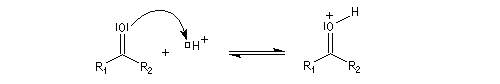

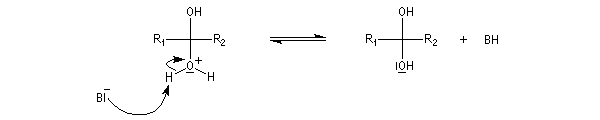
- catalyse basique.

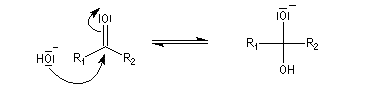
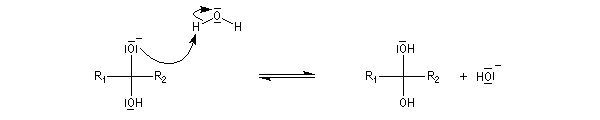
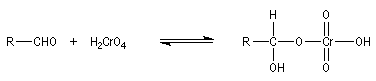
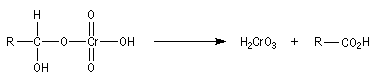
Généralités La réaction entre un composé organométallique et un aldéhyde ou une cétone est généralement conduite en deux étapes :
- le composé carbonylé, dissous dans un solvant organique comme le toluène est mis à réagir avec l'organométallique ;
- le produit d'addition est un alcoolate magnésien. Cet alcoolate est hydrolysé en milieu acide afin d'éviter la précipitation de Mg(OH)2 . L'hydrolyse conduit à l'alcool. Dans la synthèse des alcools tertiaires il faut éviter d'utiliser un milieu trop acide afin d'éviter la déshydratation de l'alcool.
Aldéhydes
La réaction entre un organomagnésien et un aldéhyde, suivie d'une hydrolyse acide, constitue une synthèse d'alcool secondaire. Voici quelques exemples.
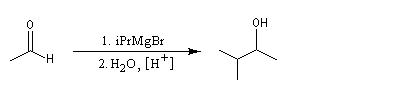
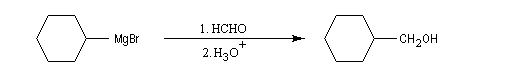

Les cétones sont moins réactives que les aldéhydes à cause d'un carbone plus encombré et moins électrophile. La réaction entre un organomagnésien et une cétone conduit à un alcool tertiaire.
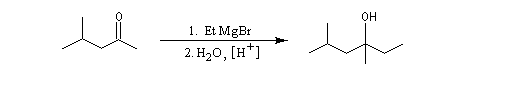
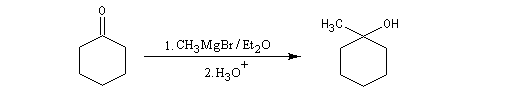
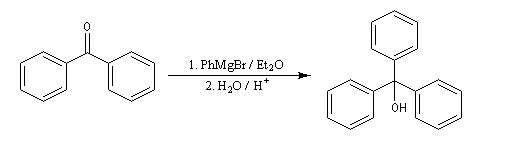
|
Composé
|
méthanal
|
aldéhydes
|
cétones
|
|
Classe d'alcool
|
primaire
|
secondaire
|
tertiaire
|
Les mécanismes de réaction entre les organomagnésiens et les composés carbonylés ne sont pas complètement élucidés. Dans certains cas, la cinétique de la réaction est en faveur d'un mécanisme dans lequel intervient deux molécules d'organomagnésien (mais une loi cinétique ne permet pas de démontrer un mécanisme).
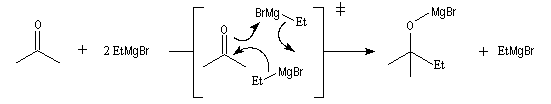
Lorsqu'on ajoute lentement du bromure de phénylmagnésium à une solution de benzophénone dans le toluène, on observe de façon transitoire une coloration rouge. On l'interprète par l'existence d'un complexe de transfert de charge qui résulte d'un transfert monoélectronique du magnésien vers la cétone.
Aspect stéréochimique La stéréochimie de l'addition d'un organométallique sur les faces diastéréotopiques d'un composé carbonylé peut être prévue en utilisant le modèle de Felkin-Anh sauf quand la présence d'un groupe jouant le rôle de base de Lewis sur le substrat entraîne un contrôle par chélation de l'addition.
Oléfination de Wittig
Introduction Le chimiste allemand G. Wittig qui est décédé en 1987, a travaillé dans de nombreux domaines de la chimie organique. Il est surtout connu pour la découverte de la réaction qui porte son nom et qui lui a valu le prix Nobel de chimie en 1979 [31] conjointement avec le chimiste américain H. C. Brown. En 1950, G. Wittig, qui était déjà l'auteur de nombreux travaux en chimie organométallique, cherchait à préparer des composés pentavalents de l'azote. L'équation de la réaction qu'il voulait réaliser s'écrit :
Inutile d'insister sur le fait qu'il n'obtint pas le produit convoité (!) mais un composé possédant une liaison carbone-azote provenant de la métallation d'une liaison C-H du substrat par le méthylithium. Les ylures d'azote étaient découverts.
"At the time we were not sure whether hydrogen bound to carbon would be proton-labile in quaternary ammonium salts. We came to this conclusion with an absurd experiment to prepare pentamethylnitrogen from tetramethylammonium salts by using the reaction of tetramethylammonium halide with methyllithium. It was confirmed experimentally that the octet principle is strictly valid for the elements of the first eight-element period. The object of synthesizing compounds with a pentacoordinate central atom was reached only when we studied the higher elements of the fifth main group - that is, phosphorus, arsenic, antimony, and bismuth." (extrait de la conférence donnée par G. Wittig à l'occasion de la remise de son prix Nobel en 1979) [68].
Tout naturellement Wittig se tourna ensuite vers le phosphore dont l'aptitude à étendre son octet est bien connue dans des composés comme PCl5. Avec un sel de phosphonium, l'équation de la réaction est :
.
Trois ans plus tard, G. Wittig et G. Geissler publiaient un article dans lequel ils décrivaient une méthode de préparation d'ylures de phosphore et leur réaction avec des composés carbonylés [44]. Y était décrite en particulier la réaction suivante qui s'effectue par simple mélange des réactifs.
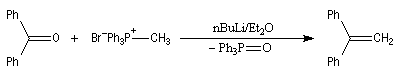
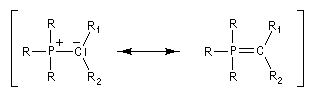
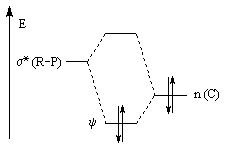
|
L'effet correspond à l'interaction entre un doublet non liant n(C) porté par l'atome de carbone négatif de
l'ylure et l'orbitale antiliante s* de la liaison R-P. Au lieu d'occuper l'orbitale n(C) les électrons occupent
l'orbitale y telle que : Cette combinaison est liante entre P et C et antiliante entre R et P. On doit donc observer une longueur de liaison P-C diminuée et une liaison R-P allongée ; résultat conforme à l'expérience. |
Les phosphines sont des bases faibles mais de bons nucléophile. On notera au passage que le pouvoir nucléophile des amines est d'autant plus élevé qu'elles sont moins substituées alors qu'on observe l'inverse chez les phosphines. On peut attribuer cette différence à la taille plus importante de l'atome de phosphore par rapport à celle de l'atome d'azote.
 |
A la température ordinaire, la triphénylphosphine se présente sous la forme d'un solide cristallin stable. Une préparation de la triphénylphosphine consiste à faire réagir un organomagnésien avec le trichlorure de phosphore. |
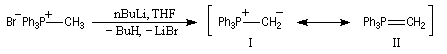
Plus généralement, on distingue schématiquement trois grandes catégories d'ylures :
- les ylures non stabilisés sont souvent des composés fortement colorés dont la préparation nécessite des bases très fortes. Le butyllithium dans l'éthoxyéthane ou le THF rigoureusement anhydre sont souvent utilisés. Rappelons que le butyllithium étant pyrophorique,
il est indispensable de manipuler ce composé sous atmosphère inerte
afin d'éviter tout risque d'incendie. Wittig utilisait volontiers le phényllithium qu'il avait préparé par la réaction d'échange métal-halogène (dans son livre : Logique de la Synthèse Organique, P. Laszlo rapporte que G. Wittig avait coutume d'appeler le
phényllithium "sa baguette de sourcier" [17].)
D'autres combinaisons de solvants et de bases peuvent être utilisés. Notamment : l'amidure de sodium dans l'ammoniac liquide, le carbanion dimsyle préparé par action de NaH sur le DMSO. Le mélange de bromure de triphénylméthylphosphonium et de NaH dans le THF absolu constitue le réactif instant ylid de Schlosser. Ces ylures non stabilisés sont préparés au moment de l'emploi au sein même du milieu réactionnel. Ils réagissent immédiatement avec l'eau et le dioxygène c'est pourquoi leur préparation exige un milieu rigoureusement anhydre, sous atmosphère inerte (diazote, argon). - Les ylures stabilisés peuvent être préparés avant l'emploi et
certains sont disponibles commercialement. Il sont compatibles avec
l'utilisation d'un milieu aqueux et une base comme l'ion hydroxyde peut
convenir.
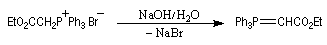
- Les ylures semi-stabilisés constituent une catégorie intermédiaire entre les deux précédentes. Le tableau suivant résume ces propriétés.
|
Ylure
|
non stabilisé
|
semi-stabilisé
|
stabilisé
|
|
Formule
|
|
|
|
|
Nature des goupes
|
-H, alkyle
|
-OR, -X, phényle, vinyle
|
-CO2Et, -SO2Ph,
-CHO, -CN |
|
Préparation
|
in situ
|
in situ
|
à part
|
Réaction avec les composés carbonylés La vitesse de la réaction augmente avec le caractère électrophile du composé carbonylé. Les aldéhydes réagissent plus vite que les cétones et il est possible d'effectuer l'oléfination de la double liaison d'un aldéhyde en présence d'un groupement oxo dans certaines conditions. Les esters de l'acide méthanoïque réagissent avec le plus simple des ylures pour conduire aux éthers d'énols. Les cétones ne réagissent que très difficilement avec les ylures disubstitués. Les alcènes tétrasubstitués ne peuvent pas être préparés valablement par cette méthode.
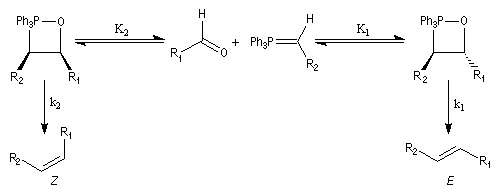
Mécanisme de la réaction de Wittig Le mécanisme de la réaction a fait l'objet de plusieurs hypothèses dès sa découverte. L'existence d'un intermédiaire appelé oxaphosphacyclobutane ou, plus communément, oxaphosphétane a été proposée par Wittig dans son article initial [44]. L'existence de cet intermédiaire a été démontrée par E. Vedejs en 1973 en utilisant la spectroscopie RMN du 31P à basse température [56]. L'expérience ainsi que les calculs théoriques militent en faveur d'une cycloaddition [2 + 2] [58].
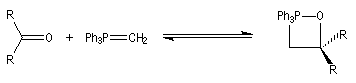
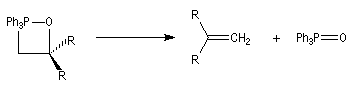
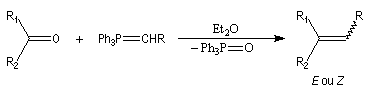
On observe expérimentalement que la décomposition des oxaphosphétanes est stéréospécifique :
- un oxaphosphétane dans lequel les groupes R1 et R2 sont en relation cis conduit à un éthylénique de configuration Z ;
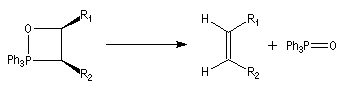
- un oxaphosphétane dans lequel les groupes R1 et R2 sont en relation trans conduit à un éthylénique de configuration E.
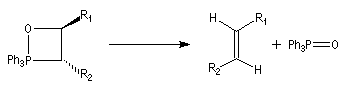
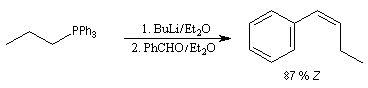
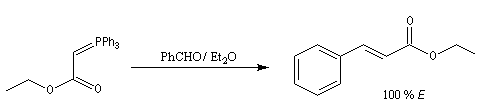
|
Ylure
|
non stabilisé
|
stabilisé
|
|
Ethylénique
|
> 90 % Z
|
> 90 % E
|
L'explication la plus classique de ces résultats expérimentaux est la suivante :
- Sous contrôle
cinétique, le produit primaire de l'addition de Wittig entre un ylure
non stabilisé, très réactif, et un composé carbonylé, doit correspondre à
un état de transition précoce. D'après le postulat de Hammond, celui-ci ressemble aux réactifs. Cet état de transition
nécessite une disposition orthogonale des liaisons réagissantes afin de
conduire à une cycloaddition [2 + 2] qui soit permise par les règles de
Woodward-Hoffmann. Dans l'approximation des orbitales frontières, l'ylure intervient par sa HO et le composé éthylénique intervient par sa BV. La géométrie de l'addition est supra pour l'ylure et antara pour le composé carbonylé (les liaisons se forment au dessus et en dessous du plan du carbonyle).
L'état de transition de plus basse énergie est celui dans lequel les groupements R1 et R2 sont aussi éloignés que possible de façon à minimiser les interactions de nature stérique.
Cet état de transition possède une certaine ressemblance avec celui rencontré dans l'addition entre un cétène et un composé éthylénique qui fournit un cyclobutane substitué. L'état de transition précédent conduit à un oxaphosphétane cis.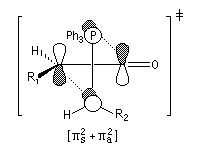
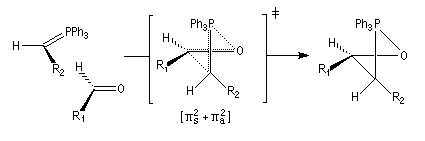 La décomposition rapide de cet oxaphosphétane fournit l'éthylénique Z.
La décomposition rapide de cet oxaphosphétane fournit l'éthylénique Z.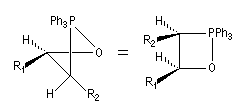
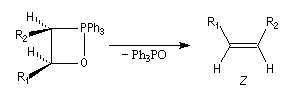
- La réaction impliquant un ylure stabilisé est vraisemblablement l'objet d'un contrôle thermodynamique. L'oxaphosphétane cis peut s'isomériser en oxaphosphétane trans, par l'intermédiaire des réactifs de départ. A l'équilibre, l'oxaphosphétane trans, plus stable que le cis, est majoritaire. Par ailleurs, le postulat de Hammond permet de prévoir que la réaction d'élimination doit être plus rapide pour l'oxaphosphétane trans que pour le cis. En effet, l'état de transition tardif ressemble au produit et les éthyléniques trans sont plus stables que les cis. On obtient donc majoritairement l'éthylénique de stéréochimie E.
Réactions en l'absence de sel de lithium En présence de sels de lithium, on constate que la sélectivité cis diminue fortement. On interprète cette dérive stéréochimique par l'intervention de l'équilibre d'isomérisation entre les oxaphosphétanes cis et trans induite par les sels de lithium. Comme la présence de lithium est une conséquence de l'utilisation de bases lithiées, il faut prendre des précautions particulières pour conserver le contrôle cinétique même en présence d'ylures non stabilisés. Les réactions de Wittig en l'absence de sel de lithium sont conduites en utilisant des bases comme NaH, le tertiobutylate de potassium (tBuOK) ou le bis(triméthylsilyl)amidure de potassium (KHMDS). La réaction suivante fait partie d'une synthèse d'un alcaloïde produit par une espèce de grenouille venimeuse [71].
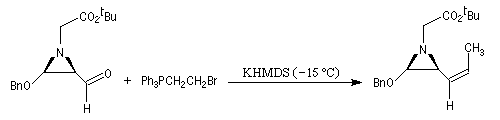
Notons qu'une bétaïne lithiée a été mise en évidence par spectroscopie RMN à basse température, durant le déroulement d'une réaction de Wittig, dans un cas particulier, assez récemment [55].
 |
L'image de gauche représente une bétaine phosphorée. Il s'agit d'un ion dipolaire ou zwitterion.
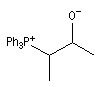 Le nom de bétaïne vient de l'analogie structurale avec celle du dérivé triméthylé du glycocolle présent dans la betterave (beta vulgaris en latin). |
- l'addition d'un organomagnésien suivie d'une hydrolyse fournit le 1-méthyl-cyclohexan-1-ol ;
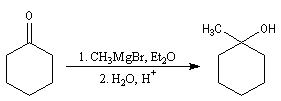
- On peut ensuite espérer obtenir le produit souhaité en
effectuant une réaction d'élimination à partir de cet alcool.
Malheureusement, le méthylène cyclohexane n'est qu'un produit très
minoritaire de cette réaction car l'élimination, conformément à la règle de Zaytzeff, fournit majoritairement le composé éthylénique le plus substitué.
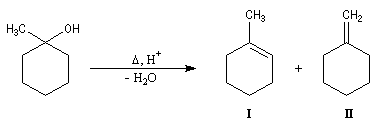
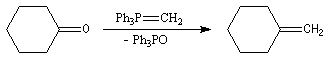
La réaction de Wittig a été utilisée comme étape clé dans de nombreuses synthèses totales de phéromones telles que la frontaline ou le bombykol.
Dans la synthèse de Bestmann du bombykol, l'ester méthylique de l'acide undec-10-énoïque subit une ozonolyse en milieu réducteur.
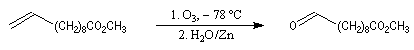
- la première réaction, qui utilise un ylure stabilisé, fournit sélectivement un produit de stéréochimie E ;
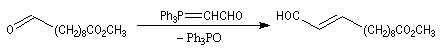
- la seconde, qui met en jeu un ylure non stabilisé, permet d'introduire la deuxième double liaison de stéréochimie Z.
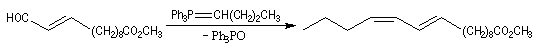
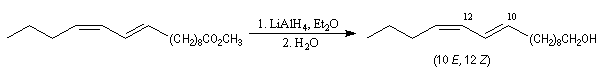
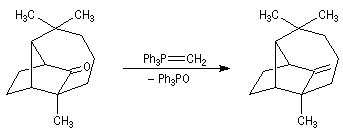
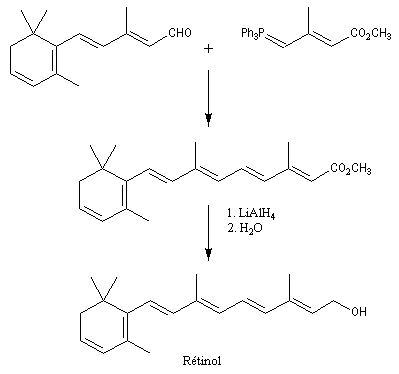
 |
Le rétinol est le précurseur biochimique d'une molécule importante dans la chimie de la vision : le rétinal. Dans l'organisme, le rétinol est transporté par une protéine dont la structure est assez curieuse et qui est appelée 1 RBP (retinol binding protein). L'isomérisation du rétinal est à la base du mécanisme de la vision. Une autre voie synthétique d'accès au rétinol est le couplage de Suzuki. |
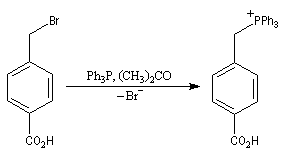
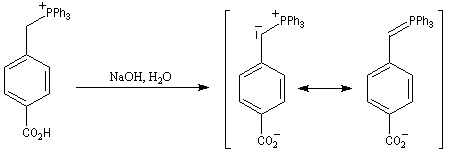
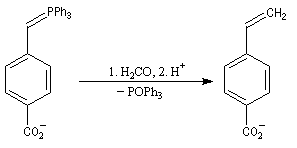
|
Groupe R
|
-Ph
|
-nC5H11
|
|
% éthylénique E
|
97
|
99
|
- l'ylure est formé par réaction entre le sel de phosphonium
et le phényllithium en présence de bromure de lithium. La réaction de ce
dernier avec
l'aldéhyde, en présence d'un excès de lithium conduit rapidement et
quantitativement à un mélange de lithiobétaïnes diastéréo-isomères.
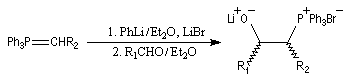
- le mélange est alors traité par un deuxième équivalent de
mélange phénylithium-bromure de lithium, ce qui conduit à la
déprotonation des lithiobétaïnes stéréoisomères pour donner un b-oxydoylure ;
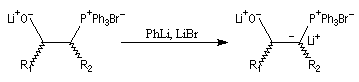
- l'étape clé de la transformation consiste à traiter le mélange par un équivalent de HCl. La protonation qui en résulte est diastéréosélective
et fournit exclusivement le mélange de diastéréo-isomères de stéréochimie like.
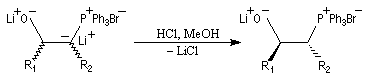
- l'étape suivante consiste à ajouter du tertiobutylate de
potassium au mélange. L'échange de cation qui en résulte conduit au sel
de
potassium de la bétaïne.
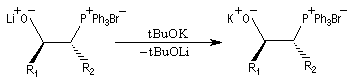
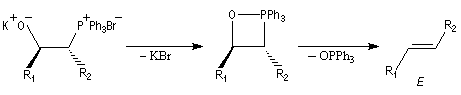
- la bétaïne privée du cation lithium stabilisateur (interaction acide dur-base dure) est instable. Après cyclisation en oxaphosphétane, on obtient un composé éthylénique de stéréochimie E.
Une application intéressante des b-oxydoylures, due à E. J. Corey, est leur utilisation dans une synthèse stéréosélective d'alcools allyliques à partir d'un aldéhyde.
Le b-oxydoylure est formé par déprotonation d'une lithiobétaïne avec le butyllithium comme cela a été vu plus haut :
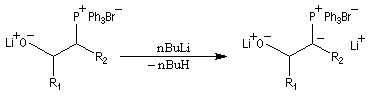
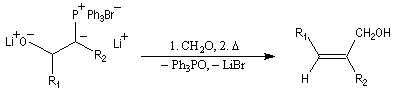
Les ylures très stabilisés ne sont généralement pas suffisamment réactifs pour donner lieu à la réaction de Wittig. Une alternative consiste à utiliser l'addition d'un carbanion phosphonate sur un composé carbonylé :
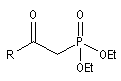
- dans la réaction de Wittig-Horner, on utilise un dialkylester de l'acide b-cétophosphonique [45] ;
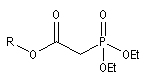
- dans celle de Wadsworth-Emmons on part d'un dialkylester de l'acide b-(alkoxycarbonyle)-phosphonique [46].
 |
 |
|
dialkylester de l'acide b-cétophosphonique
|
dialkylester de l'acide b-(alkoxycarbonyle)-phosphonique
|
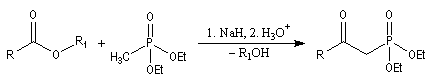
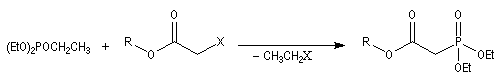
- l'alkylation du trialkylphosphite est une substitution nucléophile bimoléculaire (SN2) ;
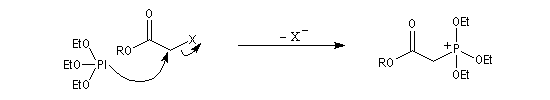
- l'ion halogénure déplace le groupe alkyle de l'intermédiaire à
la suite d'une deuxième substitution nucléophile sur l'intermédiaire ;
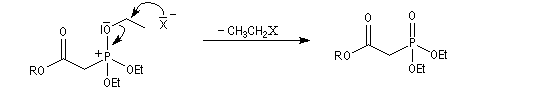
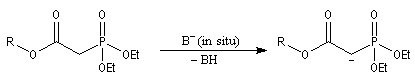
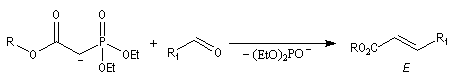
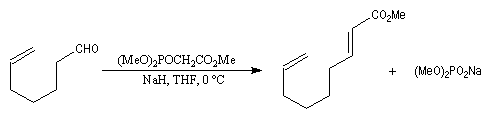
- la préparation du carbanion peut être effectuée dans des conditions douces en utilisant une base comme le DBU ;
- les phosphonates sont plus réactifs que les ylures stabilisés utilisés dans la réaction de Wittig. La réaction peut être effectuée en milieu biphasé en utilisant une catalyse par transfert de phase ;
- les carbanions phosphonates sont moins chers que les ylures ;
- le sous-produit phosphoré est soluble en milieu aqueux ce qui facilite sa séparation du milieu réactionnel en fin de réaction.
La préparation du trans-stilbène à partir du benzaldéhyde constitue un exemple de ce type de réaction.
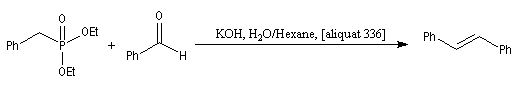
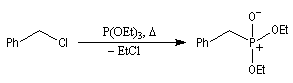
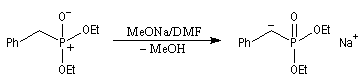
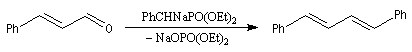
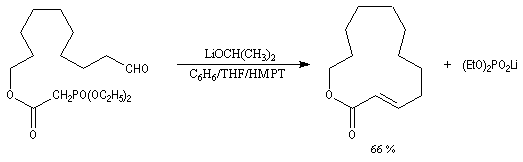
Il est possible d'obtenir de façon stéréosélective des composés éthyléniques de stéréochimie Z en utilisant la modification de Still-Genarri de la réaction de Wadsworth-Emmons. Celle-ci consiste à greffer un groupe fortement électroattracteur au carbanion phosphonate. L'origine de ce changement de stéréosélectivité reste assez obscure.
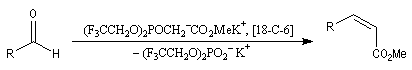
Introduction Les carbanions silylés peuvent être obtenus par métallation d'un silane par une base forte comme le butyllithium.
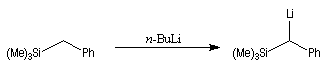
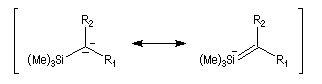
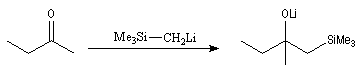
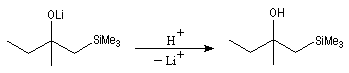
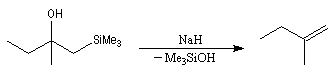
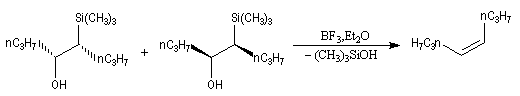
L'un des avantages de cette oléfination par rapport à celle de Wittig, tient à la facilité de séparation des produits obtenus. Contrairement à l'oxyde de phosphine formé lors de la réaction de Wittig qui peut être difficile à extraire du milieu réactionnel, les silanols sont des composés volatils aisément séparables. Aspect stéréochimique La stéréochimie de l'élimination à partir d'un b-hydroxysilane dépend de la nature du milieu dans laquelle elle est effectuée et notamment du caractère acide ou basique de ce milieu. Considérons l'exemple suivant :
- en milieu acide, on obtient le (E)-but-2-ène ;
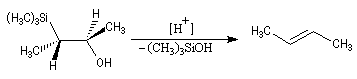
- en milieu basique, on obtient le (Z)-but-2-ène.
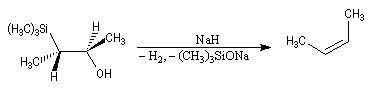
- en milieu acide, il s'agit d'une anti-élimination ;
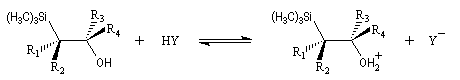
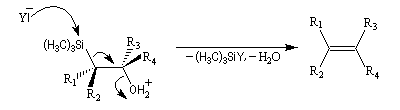
- en milieu basique, on a affaire à une syn-élimination.
La rotation autour de la liaison C-C permet à la molécule d'adopter une conformation favorable à une cyclisation.
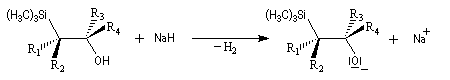
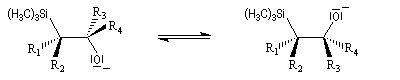
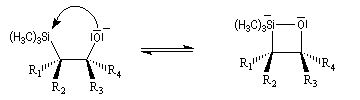
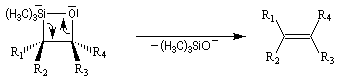
L'addition d'un carbanion a-silylé sur le butanal fournit un mélange de stéréoisomères du b-hydroxysilane ci-dessous.
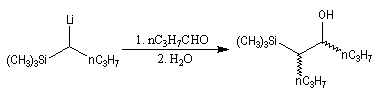
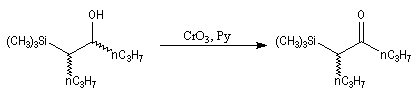
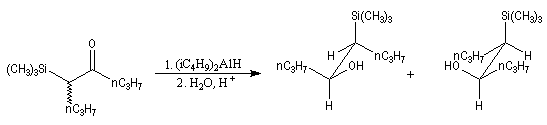
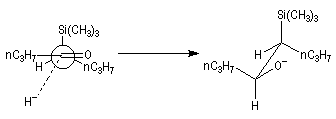
- en milieu acide l'alcène de configuration Z ;
- en milieu basique l'alcène de configuration E.
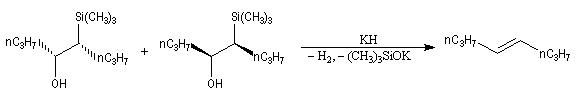
- les ylures
de sulfonium peuvent être obtenus à partir des halogénures de sulfonium.
L'halogénure lui même, est obtenu par une réaction de substitution
nucléophile.
Le sel de sulfonium (pK ~ 25) est ensuite traité par une base très forte telle que le butyllithium à - 10 °C ;
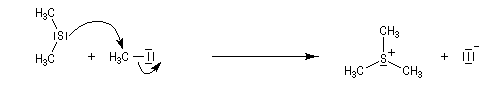
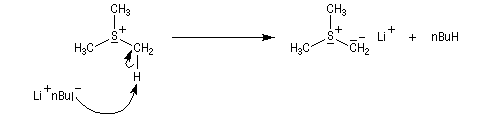 Les ylures de sulfonium sont très instables et doivent être utilisés rapidement. Ils se décomposent pour fournir un carbène (ici, un méthylène) :
Les ylures de sulfonium sont très instables et doivent être utilisés rapidement. Ils se décomposent pour fournir un carbène (ici, un méthylène) :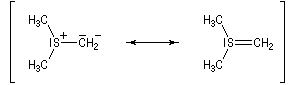
Les carbènes très instables se couplent pour donner un commposé éthylénique (ici, l'éthène).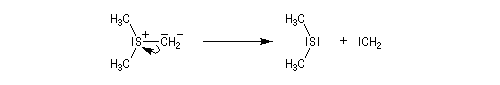
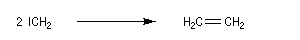
- les ylures de sulfoxonium sont obtenus selon une voie
comparable mais ils sont beaucoup plus stables et leur préparation peut
être effectuée à la température ordinaire. Un halogénure de sulfoxonium
(pK ~ 18) encore appelé réactif de Corey-Chaykowsky (en abrégé
CCR acronyme formé à partir du nom anglais : Corey-Chaykowsky Reagent)
est traité par NaH ou le carbanion dimsyle dans le DMSO.
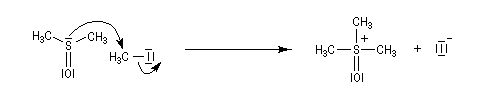
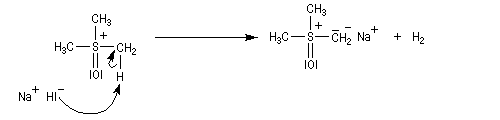
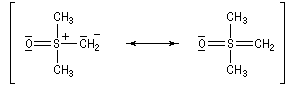
- ylure de sulfonium ;
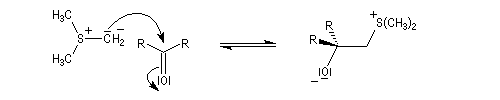
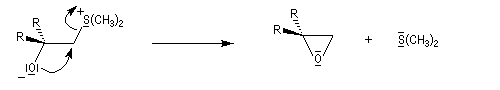
- ylure de sulfoxonium.
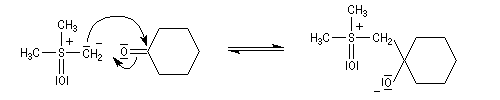
Ce dernier exemple illustre la différence de réactivité entre les ylures de sulfoxonium et les ylures de phosphore. On sait qu'avec ces derniers, on observe la formation d'un cycle à quatre chaînons appelé oxaphosphétane qui se décompose pour donner un composé éthylénique et un oxyde de triphénylphosphine Ph3PO qui possède une énergie de liaison phosphore-oxygène très grande. C'est la formation de ce composé très stable qui est à l'origine de cette orientation.
Notons que cette réaction rappelle celle de Darzens qui permet la synthèse des esters glycidiques. Aspect stéréochimique L'un des attraits de la réaction de Corey-Chaykowsky dans la préparation des époxydes, est sa stéréosélectivité :
- les ylures de sulfonium peu stables, conduisent au produit
le plus vite formé (qui est en même temps le moins stable) comportant
une liaison C-C axiale sous contrôle cinétique ;
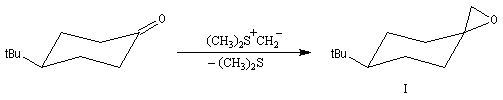
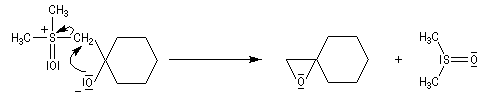
- les ylures de sulfoxonium plus stables, conduisent au produit le plus stable comportant une liaison C-C équatoriale sous contrôle thermodynamique .
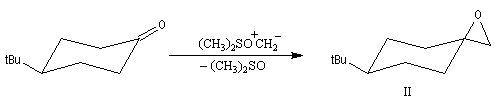
-
avec un ylure de sulfonium, on obtient un époxyde ;
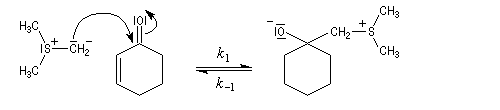 La réaction entre l'ylure et le carbonyle est quasiment irréversible. En effet la substitution nucléophile interne, rapide et irréversible, est beaucoup plus rapide que la réaction inverse de décomposition de l'intermédiaire (k2 >> k-1). L'ensemble de la transformation conduit à l'époxyde (réaction nucléophile dur, électrophile dur).
La réaction entre l'ylure et le carbonyle est quasiment irréversible. En effet la substitution nucléophile interne, rapide et irréversible, est beaucoup plus rapide que la réaction inverse de décomposition de l'intermédiaire (k2 >> k-1). L'ensemble de la transformation conduit à l'époxyde (réaction nucléophile dur, électrophile dur).
- Avec un ylure de sulfoxonium, on obtient un cyclopropane.
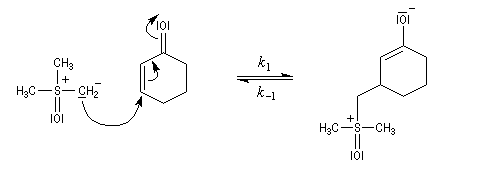
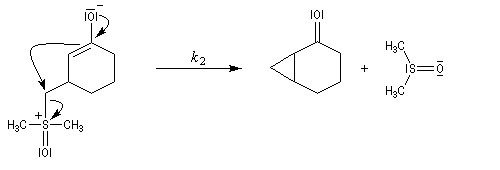
Mais l'ylure peut également réagir de façon conjuguée. L'addition conjuguée est quasiment irréversible car la substitution nucléophile ultérieure, rapide et irréversible est beaucoup plus rapide que la réaction inverse de décomposition de l'intermédiaire (k2 >> k-1). L'ensemble de la transformation conduit à un cyclopropane (interaction nucléophile mou-électrophile mou) [74].
Addition du diazométhane Le diazométhane est un ylure de méthane diazonium. La première étape de l'addition est la même qu'avec un ylure de soufre.
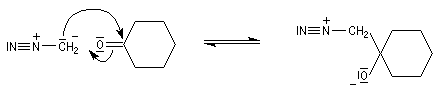
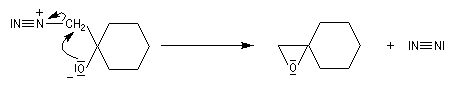
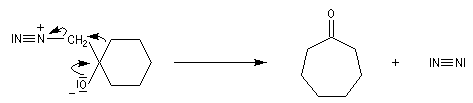
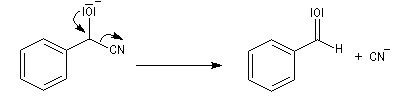
Conditions expérimentales La réaction entre HCN et un composé carbonylé conduit à un a-hydroxynitrile qu'on appelle aussi une cyanhydrine. Le cyanure d'hydrogène HCN n'étant pas nucléophile, on fait réagir le composé carbonylé avec un cyanure alcalin puis on acidifie progressivement le milieu réactionnel. Il est intéressant de noter la valeur suivante pKa (HCN/CN-) = 9,1 pour le couple acide-base correspondant.
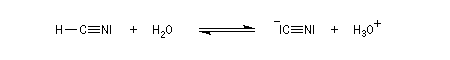
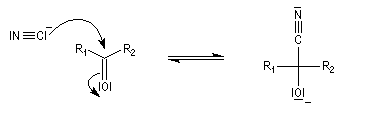
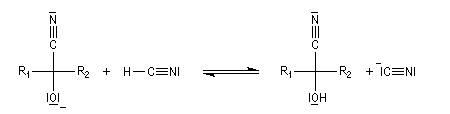
La réaction suivante concerne la synthèse du 2-hydroxy-2-phényléthanenitrile racémique plus connu sous le nom de mandélonitrile [23].

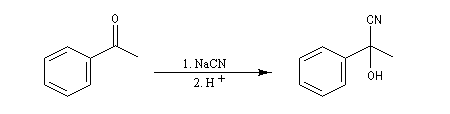
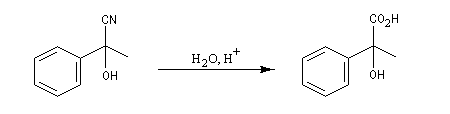
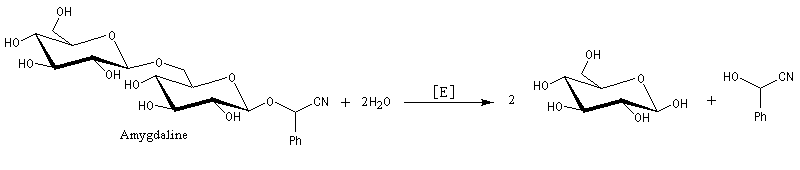
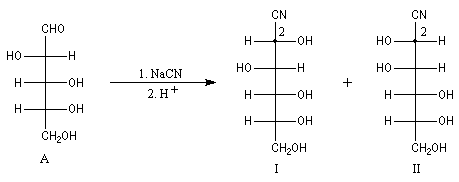
L'addition de HCN au D-arabinose (A) fournit deux a-hydroxynitriles diastéréo-isomères (I) et (II). Ces deux stéréoisomères diffèrent seulement par la configuration de l'atome de carbone C2. Ce sont des épimères.
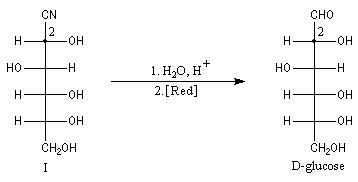

Dégradation de Wöhl Puisque la réaction de formation des cyanhydrines est renversable, on peut diminuer la longueur de la chaîne carbonée d'un sucre en traitant un a-hydroxynitrile en milieu basique. C'est le principe d'une des étapes de la réaction de dégradation de Wöhl [87].
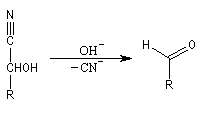
On notera qu'il existe une différence entre la synthèse de Kiliani-Fischer et la dégradation de Wöhl sur le plan stéréochimique. A partir d'un même substrat de départ, la première réaction fournit deux épimères tandis que la seconde réaction n'en fournit qu'un seul. Synthèse industrielle du PMM L'addition de HCN sur la propanone conduit au 2-hydroxy-2-méthylpropanenitrile.
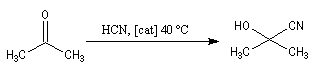
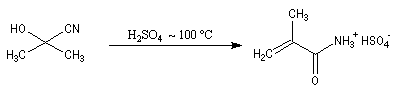
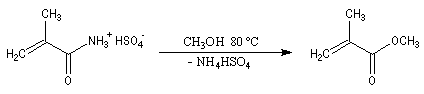
Synthèse de Strecker Les a-aminonitriles peuvent être préparés en faisant réagir un aldéhyde ou une cétone avec NaCN en présence de NH4Cl. Cette réaction, appelée synthèse de Strecker, peut être considérée comme un cas particulier de réaction de Mannich.
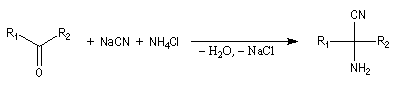
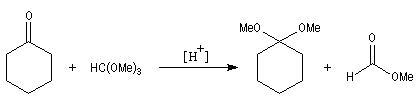
Les hexoses comme le glucose, existent principalement sous forme d'hémiacétals possédant 6 chaînons appelés glucopyranoses. Acétals et cétals La réaction entre un hémiacétal et un alcool, catalysée par un acide fournit un acétal. Du fait de leur faible réactivité, et du caractère renversable de leur réaction de formation, les acétals sont utilisés comme groupe protecteur des composés carbonylés ou des alcools. Les acétals sont stables en milieu basique. En revanche ils redonnent facilement l'alcool et le carbonylé parents par hydrolyse en milieu acide.
Les acétals sont synthétisés de façon commode par réaction d'échange avec le diméthylcétal du méthanoate de méthyle. Ce cétal d'ester est encore appelé orthoester d'où son nom courant d'orthoformiate de méthyle.
Certains acétals existent à l'état naturel. La frontaline est une phéromone d'agrégation de coléoptères appartenant à la famille des scolitidae. Parmi ces insectes, le scarabée Dendroctonus frontalis Zimmermann (Southern Pine Beetle) est l'insecte le plus destructeur de forêts de pins dans le sud des USA.
Thiols
Thiocétals
 |
L'éthanedithiol est un réactif utilisé pour la synthèse des dithiocétals.
On peut le préparer au moyen de la réaction de substitution suivante, dans laquelle les ions hydrogénosulfure sont les nucléophile :
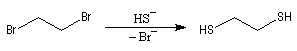 |
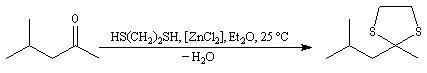
On les utilise fréquemment comme groupement protecteur des aldéhydes et des cétones. Pour débloquer le groupe carbonyle et régénérer les composés parents, divers réactifs sont disponibles. On peut par exemple traiter le thiocétal par un sel de mercure (II). La formation de HgS déplace la réaction vers la droite.
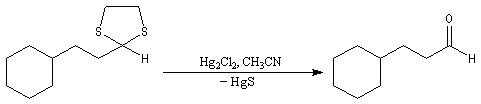
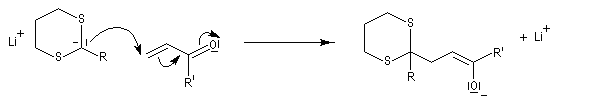
Réaction de Corey et Seebach Le but est d'effectuer la synthèse d'une cétone à partir d'un aldéhyde et d'un agent alkylant. On peut schématiser cette transformation par :
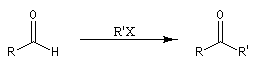
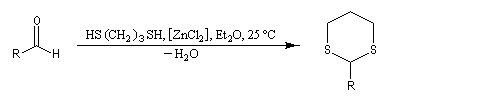
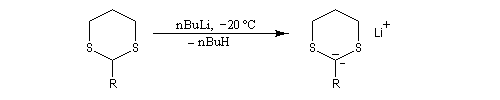
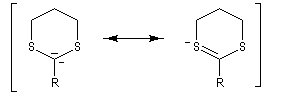
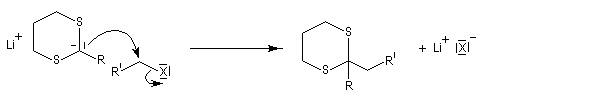
Ce carbanion est facilement alkylé au moyen d'un dérivé halogéné :
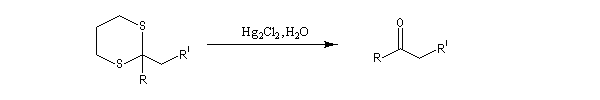
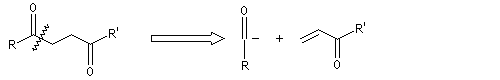
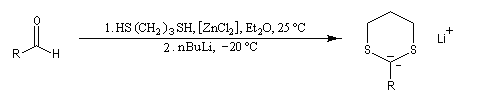
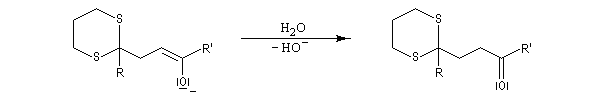
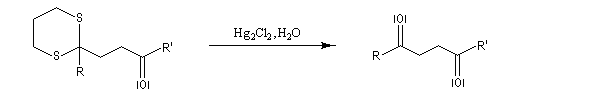
- l'ozonolyse de cyclobutènes disubstitués préparés préalablement par cycloaddition ;
- l'hydrolyse de dérivés disubstitués du furane.
- Réaction de Henry suivie d'une réaction de Nef.
Test
Ugo Schiff est un chimiste d'origine allemande qui a effectué une grande partie de sa carrière en Italie, plus précisément à Florence puis à Turin [64]. Le réactif de Schiff permet de caractériser les aldéhydes. Il est basé sur la formation d'un produit d'addition coloré entre le réactif et le composé carbonylé. La facilité avec laquelle se forme l'adduit dépend de l'encombrement de l'atome de carbone fonctionnel. C'est la raison pour laquelle le test est positif avec les aldéhydes et négatif avec la plupart des cétones sauf la propanone.
Réactions mises en jeu
 |
 |
|
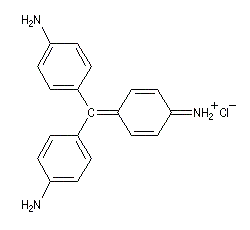
I chlorhydrate de p-rosaniline
(rose) |
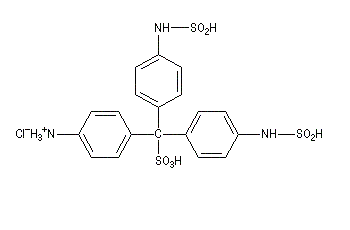
II réactif de Schiff
(incolore) |
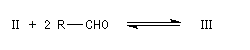
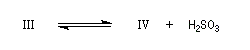
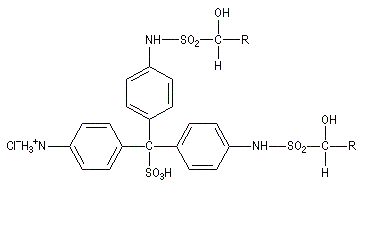 |
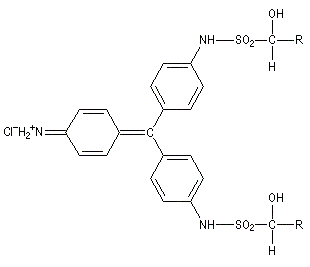 |
|
III (incolore)
|
IV (coloré)
|
- d'effectuer le test à température constante dans un mélange eau-glace ;
- d'éviter d'opérer par barbotage de gaz dans le réactif.
- la méthode de Hotchkiss-Mac Manus permet de mettre en évidence les polysaccharides dans les cellules végétales. La liaison entre deux atomes de carbone voisins portant des fonctions alcools vicinales est coupée par l'acide périodique (réaction de Malaprade). Les fonctions aldéhydes formées sont colorées par la réactif de Schiff [65].
- le test de Feulgen permet la coloration de l'ADN. Il est basé sur l'hydrolyse acide des fonctions hémiacétals du désoxyribose. Les fonctions aldéhydes libérées réagissent ensuite avec le réactif de Schiff [66].
Imines Les imines peuvent être regardées comme les analogues azotés des composés carbonylés. Leur préparation peut être réalisée par condensation entre un aldéhyde ou une cétone et une amine primaire. La réduction des imines constitue une méthode d'alkylation douce des amines primaires.
La réaction entre le carbone nucléophile d'un énol d'aldéhyde ou de cétone et un ion immonium formé in situ, s'appelle réaction de Mannich. Cette réaction est précieuse dans la synthèse des alcaloïdes possédant le squelette du tropane. Elle a été mise à profit dans la synthèse de la tropinone par R. Robinson.
Enamines Les énamines peuvent être considérées comme les analogues azotés des énols. La préparation des énamines tertiaires s'effectue en faisant réagir un composé carbonylé énolisable avec une amine secondaire.
Les énamines sont des intermédiaires très utilisés en synthèse organique. Leur acylation est une méthode de préparation des dérivés dicarbonylés 1,3.
L'une des applications les plus intéressantes des énamines en synthèse concerne l'alkylation régiosélective des cétones mise au point par G. Stork.
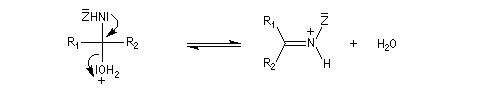
Dérivés azotés caractéristiques Cas général Nous allons symboliser le composé azoté par ZNH2. Cette écriture permet de traiter simultanément plusieurs réactions qui procèdent d'un mécanisme semblable.

|
Z
|
Réactif
|
Produit
|
Remarques
|
|
H
|
ammoniac
|
imine non substituée
|
produit non isolable
|
|
R
|
amine
|
imine substituée
|
produit isolable
|
|
NH2
|
hydrazine
|
hydrazone
|
produit solide
|
|
NHPh
|
phénylhydrazine
|
phénylhydrazone
|
produit solide
|
|
OH
|
hydroxylamine
|
oxime
|
produit solide
|
|
NH-CO-NH2
|
semi-carbazide
|
semi-carbazone
|
produit solide
|
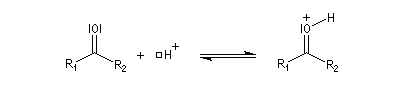
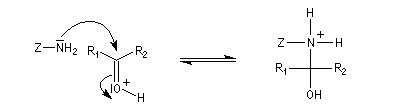
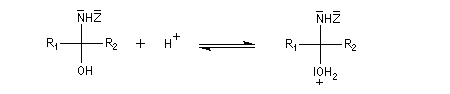
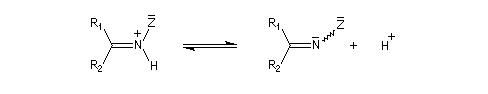
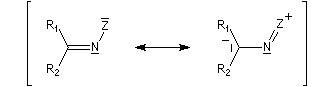
Oximes Les oximes peuvent être obtenues en faisant réagir l'hydroxylamine avec un composé carbonylé. Ce sont des composés généralement cristallisés à la température ordinaire, ce qui permet de les purifier.
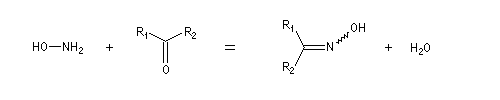
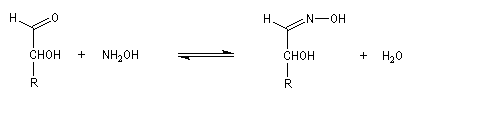
Dans la première étape, on forme l'oxime de l'ose par réaction entre ce dernier et l'hydroxylamine.
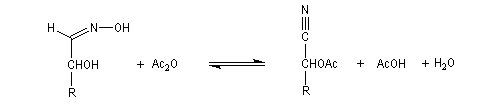
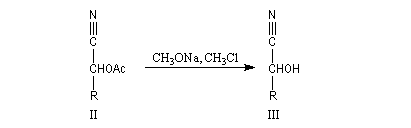
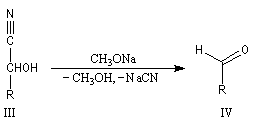
Hydrazones L'hydrazine réagit avec les composés carbonylés pour donner des hydrazones.
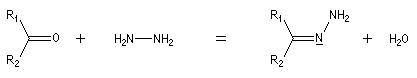
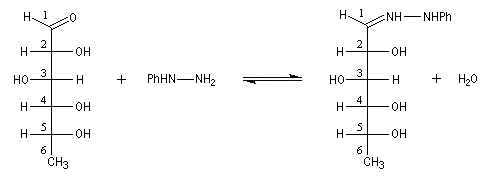
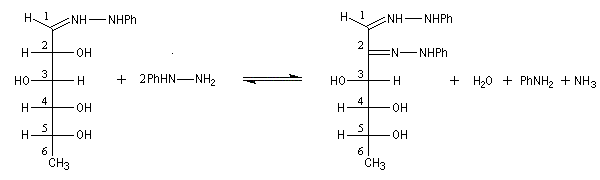
Compte tenu de la façon dont se déroule la transformation, le D-glucose, son épimère le D-mannose, mais aussi leur isomère cétonique le D-fructose, conduisent à la même phénylosazone.
La dinitro-2,4-phénylhydrazine (en abrégé 2,4-DNPH ou 2,4-DNP) est obtenue par substitution nucléophile aromatique de l'hydrazine sur le 1-chloro-2,4-dinitrobenzène. On l'utilise généralement en solution dans un mélange de méthanol et d'acide sulfurique (réactif de Brady). Ce réactif réagit encore plus facilement que la phénylhydrazine avec la plupart des aldéhydes et des cétones. On obtient une dinitro-2,4-phénylhydrazone facile à purifier. La couleur du précipité dépend de la nature du composé carbonylé de départ et s'échelonne du jaune au rouge.

 |
Quelques gouttes du composé carbonylé sont ajoutés à une
solution de 2,4-DNPH dans un mélange d'acide sulfurique et de méthanol.
Cette manière de procéder permet d'éviter
la dissolution de la 2,4-dinitrophénylhydrazone formée dans un excès de
composé carbonylé. La couleur du précipité dépend de la nature du
composé carbonylé de départ :
|
Les phénylhydrazones substituées subissent une cyclisation à chaud en présence d'un acide de Lewis pour conduire aux dérivés de l'indole selon un processus général connu sous le nom de synthèse des indoles de Fischer.
Réaction de Bamford-Stevens En présence de bases très fortes comme des alcoolates, notons que les tosylhydrazones peuvent être converties en composés éthyléniques grâce à la réaction de Bamford-Stevens [86].
Réactions d'oxydo-réduction
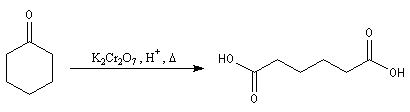
Réduction en alcool
Classe d'alcool La réduction des composés carbonylés en alcools peut être regardée formellement comme la réaction inverse de l'oxydation de ces derniers.
|
Composé carbonylé
|
méthanal
|
éthanal
|
cétones
|
|
Classe d'alcool
|
primaire
|
secondaire
|
tertiaire
|
|
Liaison
|
DrH0 (kJ.mol-1)
|
T (°C)
|
P (bar)
|
|
C=C
|
- 50
|
25
|
1
|
|
C=O
|
- 125
|
80-200
|
5-30
|
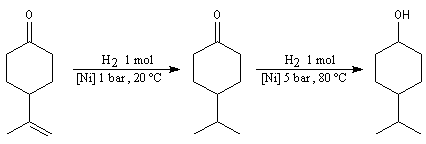

Le mécanisme fait intervenir des étapes de transfert monoélectronique et la formation d'anions-radicaux.
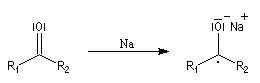
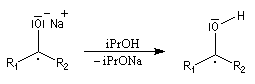
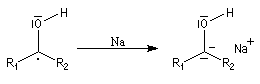
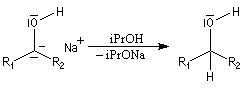
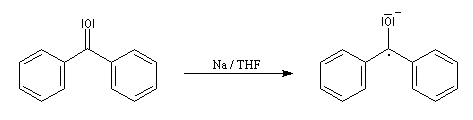
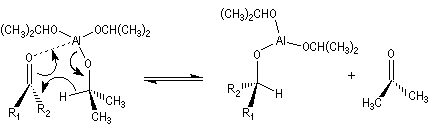
Réduction de Meerwein-Ponndorf-Verley La réduction de Meerwein-Ponndorf-Verley constitue une méthode de réduction douce des composés carbonylés. Elle procède d'un transfert d'hydrure d'un alkoxyde primaire ou secondaire. On utilise le plus souvent l'isopropylate d'aluminium qui est commercial. L'atome d'aluminium est un acide de Lewis qui permet la formation d'un complexe avec l'atome d'oxygène du carbonylé ce qui favorise la formation d'un état de transition à 6 centres de type Zimmerman-Traxler [70].
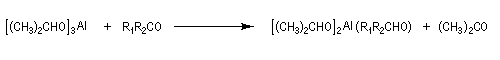
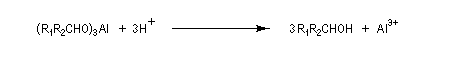
Réduction par l'alpine borane
L'alpine borane (réactif de Midland) ou le chloroborane de Brown sont des réactifs chiraux appartenant à la famille des hydrures de bore neutres (tout comme BH3 ou le DIBAL pour les hydrures d'aluminium). Ils permettent des réductions énantiosélectives de composés carbonylés. Ces réactions sont abordées dans le chapitre consacré à la stéréochimie dynamique.
Réduction de Corey-Bakshi-Shibata
Il s'agit de la réduction en alcools par le monoborane dans le THF de composés carbonylés en présence d'une oxazaborolidine comme catalyseur. Si le substrat de départ possède un groupe carbonyle dont les faces sont énantiotopiques, on obtient préférentiellement l'un des deux énantiomères possibles. Il s'agit donc d'une catalyse énantiosélective. Le temps de réaction est court à la température ordinaire. Le rendement et l'excès énantiomérique sont généralement très bons.
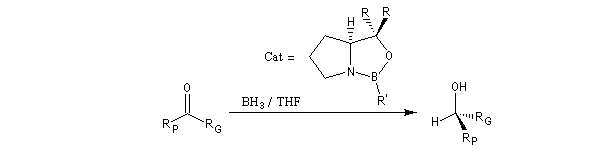
R : Ph, b-naphtyl
R' : H, Me, nBu.
La réaction a été mise à profit pour réaliser le dédoublement cinétique de mélanges racémiques.
[84] et [90] Réduction duplicative
Couplage pinacolique Lorsque les conditions sont réunies pour que les radicaux aient une durée de vie suffisante, on peut observer un couplage. Après hydrolyse, on obtient un glycol bitertiaire encore appelé pinacol. Le magnésium est efficace dans ce genre de réaction car il permet le rapprochement de deux molécules dans l'état de transition.
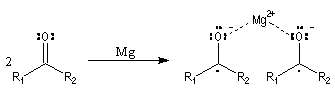
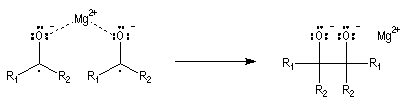
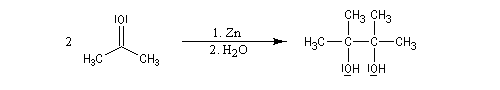
Elle a été mise à profit dans plusieurs synthèses récentes de composés complexes. L'exemple suivant concerne une étape de la synthèse du taxol par K. C. Nicolaou.
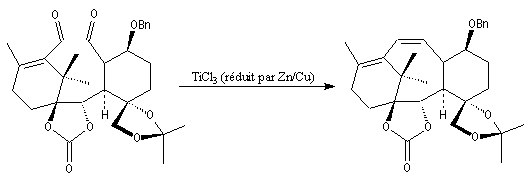
Un autre exemple concerne le couplage de deux molécules d'adamentanone conduisant à l'adamentylidèneadamentane [76].
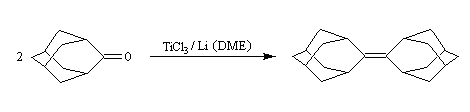
Désulfuration des thiocétals Une réaction extrêmement utile pour réduire le groupe carbonyle en méthylène fait intervenir un thiocétal comme intermédiaire. Ce dernier est préparé en traitant le composé carbonylé par l'éthanedithiol en présence d'un acide de Lewis.
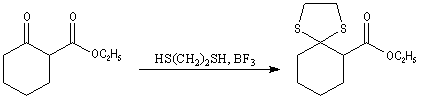
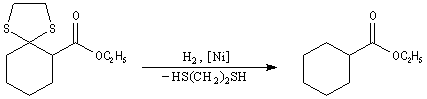
Réaction de Clemmensen La réaction consiste à traiter le composé carbonylé par un amalgame de zinc en milieu acide, à chaud. Les électrons sont fournis par le métal. L'acide joue le rôle de réactif protogène.
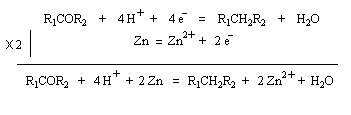
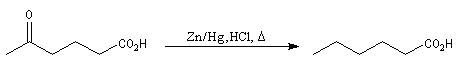
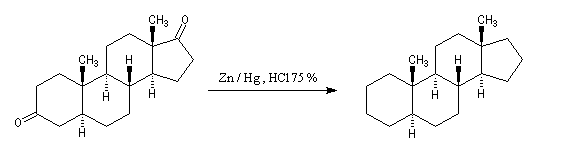
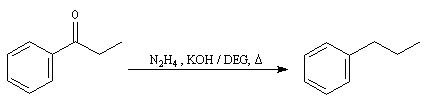
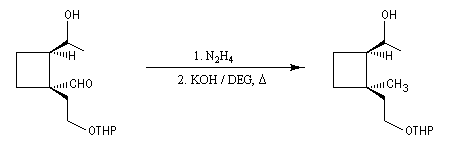
- formation d'une hydrazone.
- la base arrache un atome d'hydrogène ;
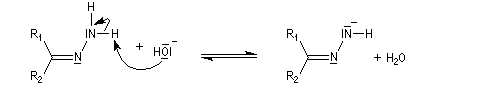
- formation d'un carbanion ;
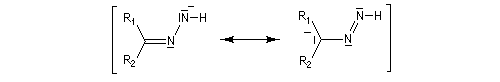
- régénération de la base ;
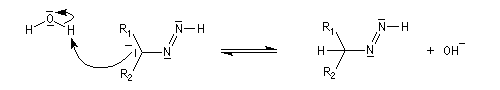
- la base arrache un nouvel atome d'hydrogène porté par l'azote ;
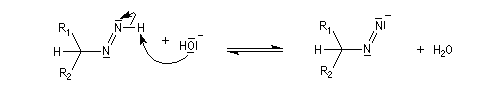
- formation d'un carbanion avec expulsion d'une molécule de diazote ;
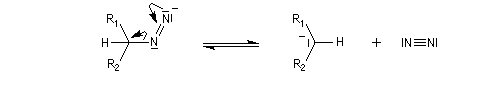
- la base est régénérée.
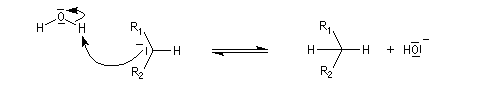
Oxydation
Aldéhydes Les aldéhydes sont des composés très réducteurs. Ils peuvent être oxydés par des oxydants doux. Les réactions suivantes utilisent des complexes d'ions métalliques en milieu basique. Elles sont surtout utilisées comme test analytique dans la chimie des sucres.
- Le réactif de Fehling est une solution
basique d'un complexe du Cu (II) avec les ions tartrate. Le ligand
masque les ions Cu (II) vis à vis des ions hydroxyde. En leur absence il
se formerait l'hydroxyde à un tel pH. Un produit de la réaction est
l'oxyde de cuivre (I). La réaction est quantitative et a été utilisée
pour effectuer le dosage des sucres réducteurs comme le glucose.

- Le réactif de Tollens est une solution d'un complexe amminé de
Ag (I) en milieu basique. Cette fois l'argent (I) est masqué vis à vis
des ions hydroxyde par le ligand ammoniac. La réaction se traduit par la
formation d'argent métallique. C'est une méthode
d'argenture des miroirs.
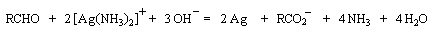
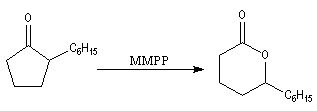
Oxydation de Baeyer-Villiger En présence d'acide métachloroperoxybenzoïque (m-CPBA) ou d'un mélange d'eau oxygénée et d'acide éthanoïque, la butan-2-one conduit à l'éthanoate d'éthyle.

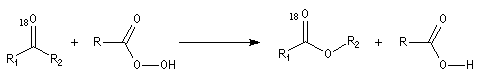
- addition entre le peroxyacide et le composé carbonylé avec formation d'un intermédiaire ;
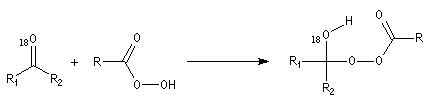
- réaction de transposition de l'intermédiaire avec migration
d'un groupe carboné sur un atome d'oxygène appauvri en électrons
conduisant à l'ester.
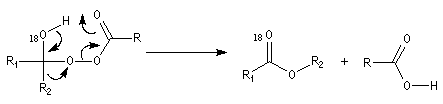
t-alkyle > s-alkyle > phényle > n-alkyle > CH3
Du point de vue stéréochimique, lorsque le problème se pose,
l'insertion de l'atome d'oxygène s'effectue avec une rétention complète
de la configuration. C'est la raison pour laquelle la réaction de
Baeyer-Villiger
a souvent été utilisée comme étape clé dans la synthèse de molécules
complexes ; en série bicyclique notamment. La réaction suivante est une
étape de la synthèse de la prostaglandine
PGF2a par E. J. Corey en 1970.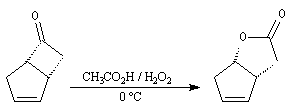
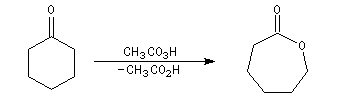
Réactions au voisinage du carbonyle
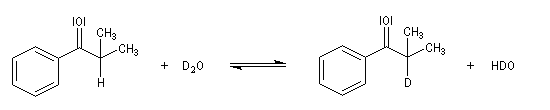
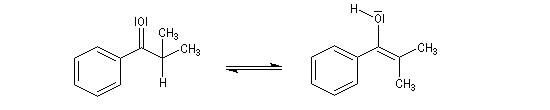
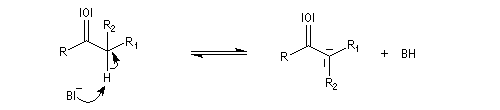
Tautomérie céto-énolique Résultats expérimentaux Réalisées à partir du même composé carbonylé comme substrat, on constate expérimentalement que les réactions suivantes s'effectuent à la même vitesse.
- Incorporation de deutérium.
Cette transformation peut être suivie grâce à la spectroscopie de RMN du proton car le deutérium ne résonne pas à la même fréquence que le proton.
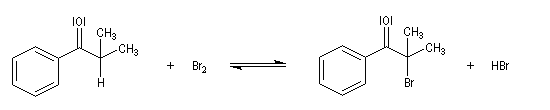
- Bromation.
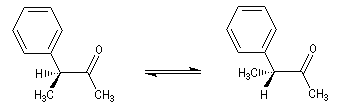
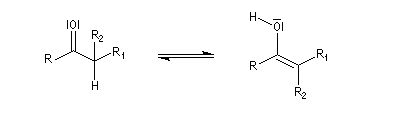
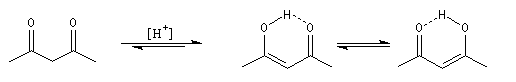
Plusieurs méthodes parmi lesquelles la spectroscopie dans l'uv - visible et la spectroscopie de RMN permettent de déterminer le pourcentage d'énol présent à l'équilibre. Le spectre ci-dessous est celui de la pentan-2,4-dione.
On note la présence de deux familles de pics de déplacements chimiques différents :
- ceux relatifs à la dicétone non énolisée (a) ;
- ceux relatifs à l'énol de la dicétone (b).
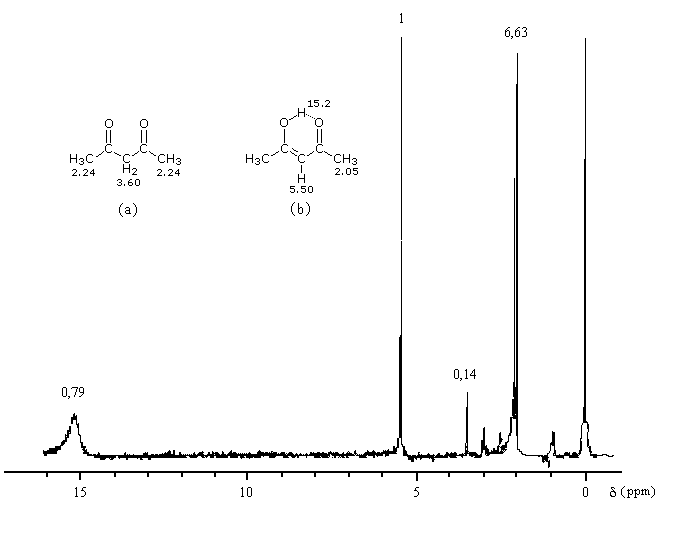
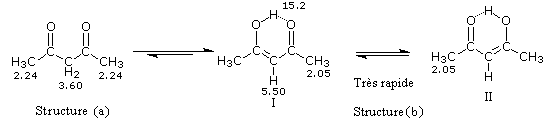
- % (a) = k´2´0,14
- % (b) = k´1´1


Taux d'énolisation Le tableau ci-dessous regroupe quelques valeurs expérimentales. Certaines particularités structurales peuvent expliquer un pourcentage d'énol élevé.
|
Composé carbonylé
|
% d'énol à l'équilibre
|
|
éthanal
|
10-4
|
|
propanone
|
10-6
|
|
pentane-2,4-dione
|
76
|
|
cyclohexane-1,2-dione
|
100
|
|
cyclohex-2,4-diène-1-one
|
100
|
- les carbonylés ordinaires sont peu énolisés. Les aldéhydes le sont davantage que les cétones ;
- chez les composés dicarbonylés l'énol peut être majoritaire.

Rappelons que : a < 0 et b < 0.
|
aC
|
aO
|
bC-O
|
|
a
|
a + 2,1 b
|
0,66 b
|
|
Orbitale moléculaire
|
Energie
|
|
j 3 = - 0,67 c 1 + 0,72 c 2 - 0,15 c 3
|
E3 = a - 1,07 b
|
|
j 2 = 0,72 c 1 + 0,61 c 2 - 0,32 c 3
|
E2 = a + 0,84 b
|
|
j 1 = 0,14 c 1 + 0,33 c 2 + 0,93 c 3
|
E1 = a + 2,32 b
|
L'énol a une réactivité nucléophile via la plus haute orbitale moléculaire occupée. La réaction avec un électrophile s'effectue préférentiellement au niveau de l'atome qui possède le plus grand coefficient dans cette orbitale. Il s'agit donc de l'atome de carbone 1.
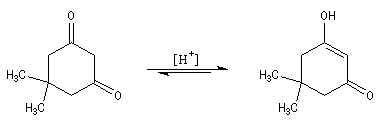
Une conséquence de la tautomérie céto-énolique : l'épimérisation en C2 des aldoses En milieu basique, le D-glucose ou (2R, 3S, 4R, 5R)- 2, 3, 4, 5, 6-pentahydroxyhexanal, et le D-mannose, deux aldohexoses dont la stéréochimie diffère par la configuration absolue de l'atome de carbone C2, sont susceptibles de s'interconvertir. La clé de cette épimérisation est la tautomérie céto-énolique qui fait intervenir comme intermédiaire un ène-diol.
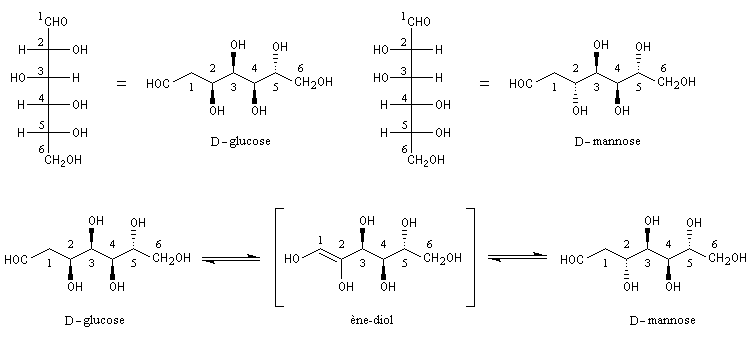
|
Composé
|
pKa (couple)
|
|
2-méthylpropène
|
45
|
|
propanone
|
25
|
|
acétophénone
|
16
|
|
pentane-2, 4-dione
|
9
|
Les atomes d'hydrogène en a d'autres fonctions sont susceptibles d'être arrachés par des bases. On obtient ainsi des ions énolate ou apparentés à partir des fonctions suivantes :
La chimie des énolates a une grande importance en synthèse car elle permet la création de nouvelles liaisons carbone-carbone.
Orbitales moléculaires de l'ion éthénolate Formellement, un ion énolate ressemble beaucoup à un système allylique. Chaque atome de carbone apporte un électron tandis que l'atome d'oxygène en fournit deux.

|
aC
|
aO
|
bC-O
|
|
a
|
a + 0,97 b
|
1,06 b
|
|
Orbitale moléculaire
|
Energie
|
|
j 3 = - 0,58 c 1 + 0,74 c 2 - 0,35 c 3
|
E3 = a - 1,28 b
|
|
j 2 = - 0,75 c 1 - 0,30 c 2 + 0,60 c 3
|
E2 = a + 0,41 b
|
|
j 1 = 0,33 c 1 + 0,60 c 2 + 0,73 c 3
|
E1 = a + 1,84 b
|
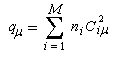
ni est le nombre d'électrons décrit par l'orbitale considérée. La somme est étendue aux M orbitales occupées.
La charge nette de l'atome m est la différence entre le nombre m d'électrons apporté au système par cet atome et sa charge électronique :
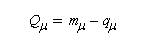
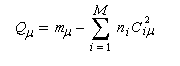
|
Q1
|
Q2
|
Q3
|
|
-0,33
|
0,09
|
-0,76
|
- les électrophiles durs réagiront de préférence au niveau de l'atome d'oxygène qui possède la charge la plus élevée ;
- les électrophiles mous réagiront de préférence au niveau de l'atome de carbone.
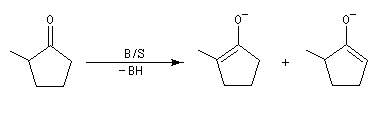
- l'énolate (II) est obtenu par déprotonation de l'atome d'hydrogène qui possède l'acidité cinétique la plus grande. Avec une base suffisamment encombrée, la vitesse de déprotonation de la cétone est la plus grande du côté le plus dégagé. Cet énolate qui est le plus vite formé sous contrôle cinétique est appelé énolate cinétique ;
- l'énolate (I), qui comporte la double liaison la plus substituée, est le plus stable. Même s'il est formé plus lentement que II, il est majoritaire quand la réaction est conduite de façon à ce qu'un équilibre entre I et II soit possible c'est à dire dans les conditions d'un contrôle thermodynamique. On l'appelle énolate thermodynamique.
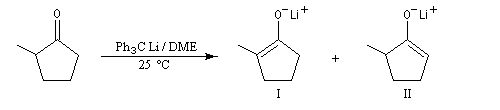
|
Conditions de la réaction
|
I
|
II
|
|
Cétone additionnée à un excès de base
|
28
|
72
|
|
Excès de cétone additionnée à la base
|
78
|
22
|
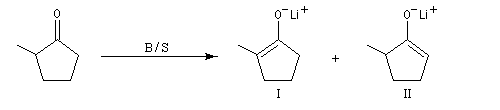
|
Base (B) / Solvant (S)
|
I
|
II
|
|
LDA/DME
|
1
|
99
|
|
Et3N/DMF
|
78
|
22
|
Ethers d'énols silylés
Un moyen pour piéger l'énolate cinétique consiste à le faire réagir avec le chlorure de triméthylsilyle le rendement de la réaction suivante est de 95 %.
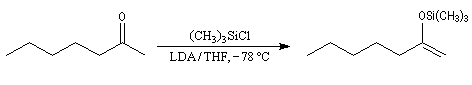
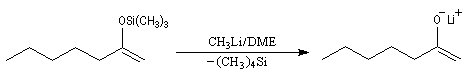
- Le solvant.
A la suite des travaux de N. Kornblum, on s'est rendu compte que l'influence du solvant est souvent déterminante. Considérons la réaction suivante choisie comme modèle :
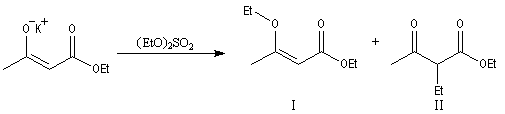
|
Produit
|
I
|
II
|
|
HMPT
|
83
|
15
|
|
tBuOH
|
0
|
94
|
Dans un solvant protique comme tBuOH, l'atome d'oxygène est lié au solvant par des liaisons hydrogène. La C-alkylation est majoritaire.
- Le contre-ion.
Les ions durs tels que Li+, Mg2+ sont fortement associés à l'oxygène négatif qui est une base dure. Il favorisent l'alkylation au niveau du carbone. Les cations mous, gros et polarisables, comme N(Bu)4+ sont moins collants et favorisent l'alkylation au niveau de l'oxygène. L'utilisation d'éthers-couronnes ou de cryptands permet dans certains cas de favoriser la O-alkylation en augmentant la dissociation des paires d'ions.
- Le nucléofuge.
L'iodométhane dont le nucléofuge, l'ion iodure, est un ion mou, réagit principalement au niveau de l'atome de carbone. En revanche les sulfates et les sulfonates sont des espèces dures. Les sulfates d'alkyle réagissent de préférence au niveau de l'atome d'oxygène.
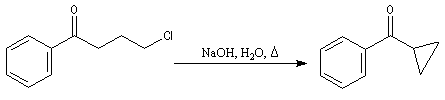
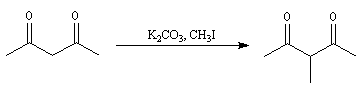
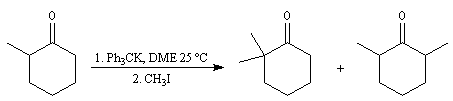
- L'énolate souhaité est préformé puis piégé sous forme d'éther d'énol silylé.
- On peut réaliser la réaction d'alkylation d'une énamine préparée à partir de la cétone et d'une amine secondaire (méthode de G. Stork).
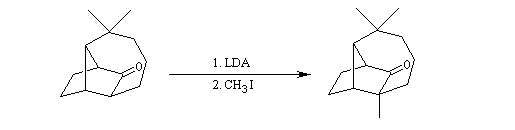
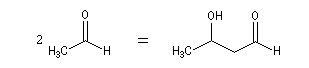
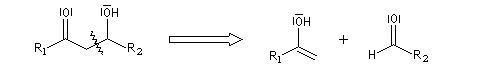
Qu'il s'agisse d'un aldol ou d'un cétol, le groupe OH est séparé du groupe carbonyle par trois liaisons simples. Voici un exemple de rétrosynthèse.
-
en milieu basique, il y a formation d'un ion énolate beaucoup plus nucléophile que l'énol ;
- l'ion énolate réagit sur l'aldéhyde ;
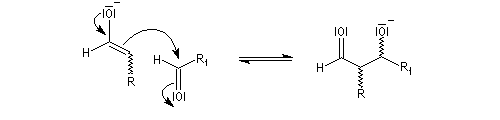
- l'ion OH- est régénéré.
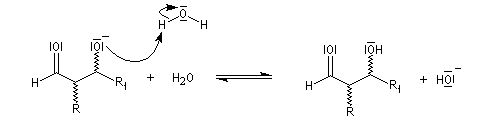

- l'ion énolate réagit sur l'aldéhyde ;
- en milieu acide, le carbonyle est protoné ;
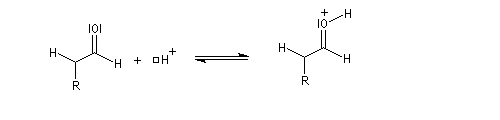
-
l'énol réagit avec le carbonyle activé pour lequel le caractère électrophile du carbone est exalté ;
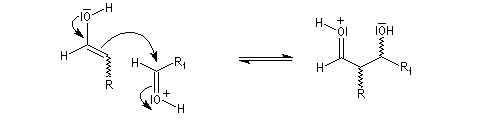
- l'ion H+ est régénéré.
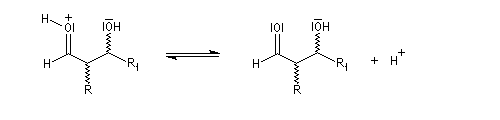
-
l'énol réagit avec le carbonyle activé pour lequel le caractère électrophile du carbone est exalté ;
- en milieu acide, le groupe OH est protoné ;
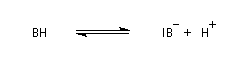
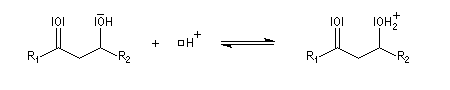
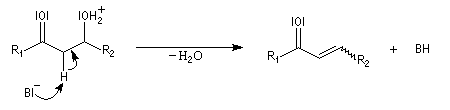
- en milieu basique, l'arrachage de l'atome d'hydrogène précède la formation d'un carbanions possédant une certaine stabilité car c'est un système conjugué.
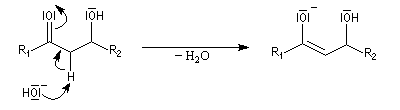 L'étape cinétiquement déterminante est l'élimination de l'ion hydroxyde, mauvais nucléofuge, à partir du carbanion. Cette étape est monomoléculaire. Il s'agit d'une élimination de type E1Cb (1 pour monomoléculaire et Cb pour base conjuguée).
L'étape cinétiquement déterminante est l'élimination de l'ion hydroxyde, mauvais nucléofuge, à partir du carbanion. Cette étape est monomoléculaire. Il s'agit d'une élimination de type E1Cb (1 pour monomoléculaire et Cb pour base conjuguée).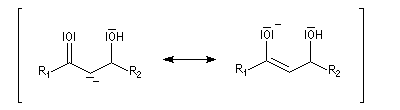
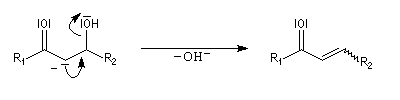
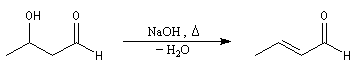
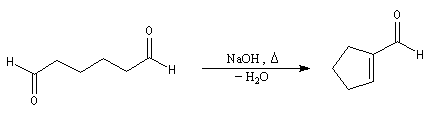
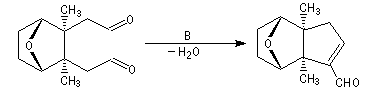
Cétolisation La réaction de cétolisation est une réaction d'addition entre deux cétones. Elle conduit à un équilibre défavorable au produit appelé cétol. On peut préparer un cétol en utilisant un extracteur de Soxhlet. Le produit est extrait au fur et à mesure de sa formation au moyen d'un système de vidange, ce qui évite sa décomposition, tandis que le réactif, plus volatil (masse molaire plus faible et pas d'association par liaison hydrogène), est recyclé par chauffage à reflux au fur et à mesure que la réaction progresse. Beaucoup d'auteurs, notamment les anglosaxons, utilisent le terme aldolisation dans un sens générique qui englobe aldolisation au sens strict et cétolisation.
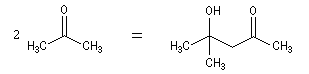
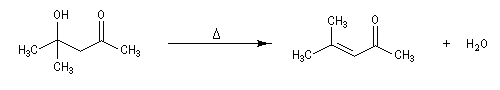
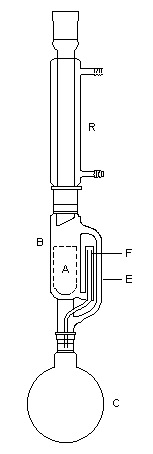
|

|
La réaction peut être effectuée entre deux cétones différentes. La préparation de la tétraphénylcyclopentadiénone (tétracyclone) se fait très facilement en mettant les réactifs à reflux dans l'éthanol en présence d'une base forte. La force motrice est la formation d'un produit très conjugué [77].

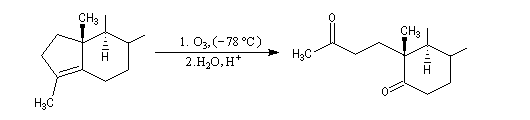
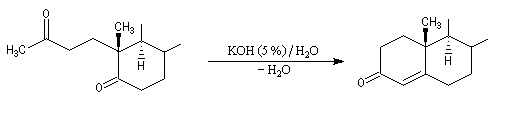
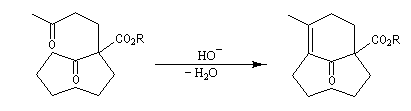
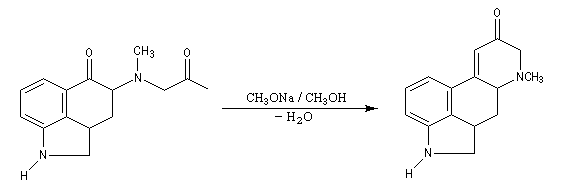
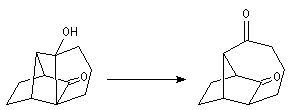
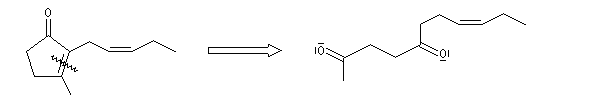
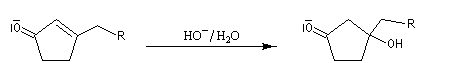
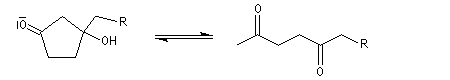
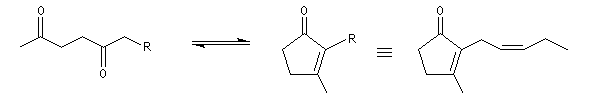
Associée à l'addition de Michaël, la condensation aldol est à la base d'une méthode générale de synthèse de cycles appelée annélation de Robinson.
Aldolisation croisée L'aldolisation croisée met en jeu un aldéhyde et une cétone. Elle conduit à un bilan simple lorsque l'un des composés n'est pas énolisable car il n'y a pas d'ambiguité sur le produit final.
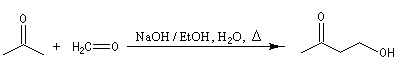
Condensation de Claisen-Schmidt
La réaction de Claisen-Schmidt constitue un cas particulier d'aldolisation croisée entre une cétone et un aldéhyde aromatique. La déshydratation de l'aldol intervient spontanément du fait de la grande stabilité du produit final fortement conjugué. L'exemple suivant concerne la préparation du 1,5-diphénylpenta-1,4-dién-3-one ou dibenzylidène acétone (en abrégé : dba). L'élimination fournit la liaison éthylénique la plus stable de stéréochimie E. Une réaction analogue peut être facilement réalisée en remplaçant le benzaldéhyde par la vanilline.
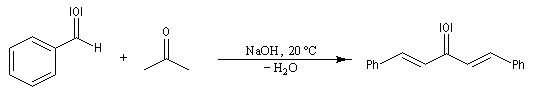
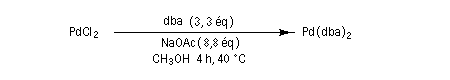
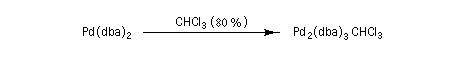
Ce complexe est utilisé dans les réactions de couplage croisé catalysées par le palladium, telles que la réaction de Heck, la réaction de Négishi et la réaction de Suzuki. Diastéréosélectivité dans la réaction aldol La condensation aldolique constitue une méthode extrêmement importante pour créer de nouvelles liaisons carbone-carbone. La méthode générale de contrôle de la nature des produits dans la condensation aldol consiste à préformer l'énolate qui doit réagir.
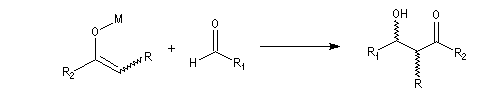
Puisqu'il existe deux configurations pour l'énolate et que le composé carbonylé comporte en général deux faces prochirales, cela conduit dans le cas général à la formation de deux centres chiraux.
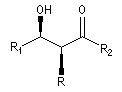 |
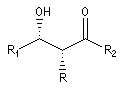 |
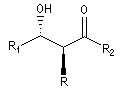 |
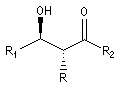 |
|
syn
|
syn
|
anti
|
anti
|
- contrôle de la stéréochimie relative, syn ou anti des centres chiraux ;
- contrôle de la configuration absolue de ces mêmes centres.
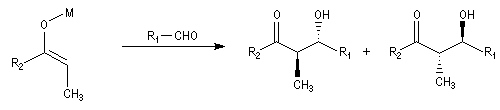
- à partir d'un énolate Z on obtient le couple d'aldols syn (érythro) ;
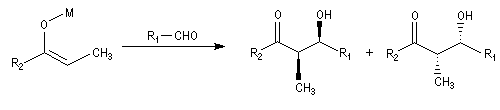
- à partir d'un énolate E on obtient le couple d'aldols anti (thréo).
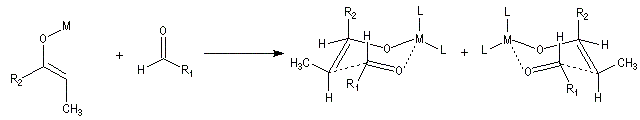
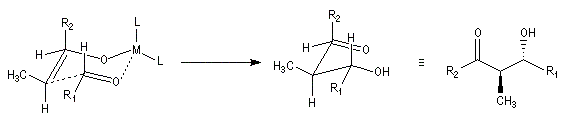
Ces résultats peuvent être rationnalisés en utilisant le modèle d'état de transition de Zimmerman-Traxler [70]. Dans ce modèle d'état de transition "fortement lié", les atomes adoptent une conformation chaise.
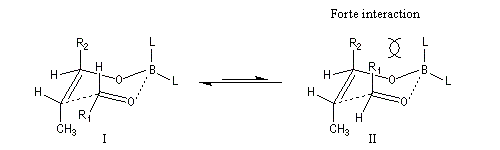
Commençons par considérer le cas d'un énolate Z. Deux états de transition diastéréoisomères sont possibles notés ci-dessous (I) et (II).

 |
 |
| L'état de transition (I) favorisé : pas d'interaction diaxiales entre les groupes les groupes R1 et R2 | L'état de transition (II) défavorisé : interactions diaxiales entre les groupes les groupes R1 et R2. |
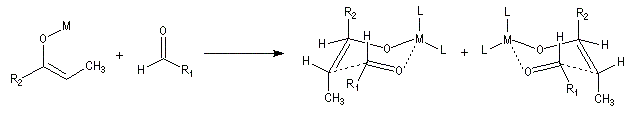
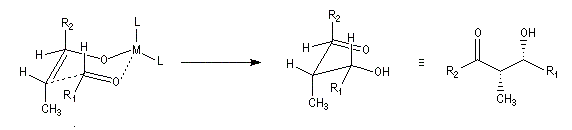
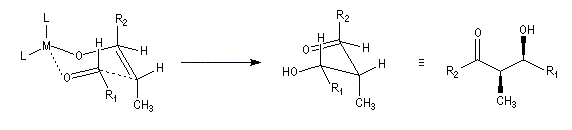
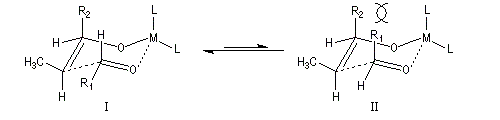
 |
 |
| Etat de transition (I) favorisé : pas d'interaction diaxiales entre les groupes R1 et R2 | Etat de transition (II) défavorisé : interactions diaxiales entre les groupes R1 et R2 |
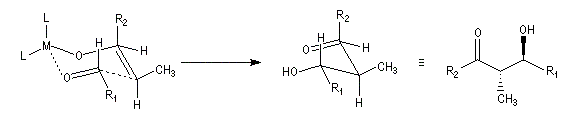
On notera que dans les réactions précédentes, il n'y a aucun contrôle de la configuration absolue puisque les formations d'états de transition énantiomères sont équiprobables. On obtient donc un mélange racémique d'aldols énantiomères.
Obtention d'énolates de lithium Z et E
Puisque la stéréosélectivité de la réaction aldol dépend de la stéréochimie de l'énolate, l'obtention d'énolates Z ou E constitue un problème important. Le rapport Z/E dépend de plusieurs facteurs :
- la nature de la base ;
- la nature du substrat ;
- le solvant.
- Les amidures de lithium peuvent conduire aux énolates Z ou E avec une diastéréosélectivité plus ou moins élevée. Les résultats ci-dessous sont obtenus dans le THF.
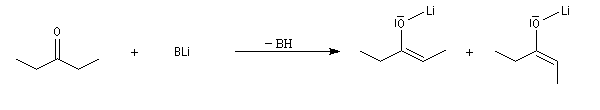
|
Base
|
Enolate Z
|
Enolate E
|
|
LHMDS
|
66
|
34
|
|
LDA
|
33
|
77
|
|
LTMP
|
14
|
86
|
- LHMDS donne très majoritairement l'énolate Z ;
- LDA donne majoritairement l'énolate E et une proportion non négligeable de Z ;
- LTMP, base très encombrée, donne très majoritairement l'énolate E ;
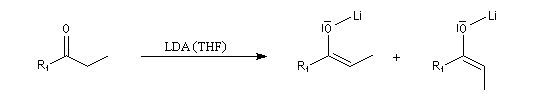
|
R1
|
Enolate Z
|
Enolate E
|
|
-OtBu
|
5
|
95
|
|
-Et
|
23
|
77
|
|
-CH(CH3)2
|
60
|
40
|
|
-C(CH3)3
|
100
|
0
|
- une base très encombrée défavorise la formation de l'énolate Z.
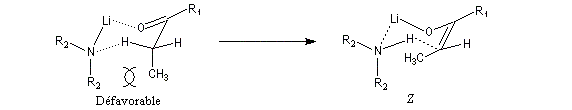
- un groupe R1 volumineux dans le substrat défavorise la formation de l'énolate E ;
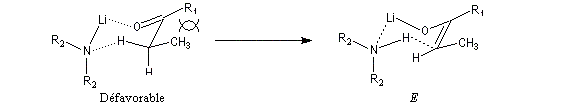
|
Base
|
Enolate Z
|
Enolate E
|
|
LTMP
|
14
|
86
|
|
LTMP (HMPA)
|
92
|
8
|
Les énolates de bore peuvent en général être obtenus avec de très bons rendements et d'excellentes diastéréosélectivités [67].
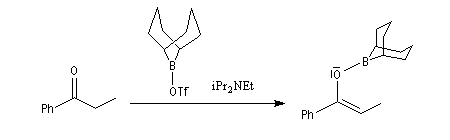
- La préparation des énolates de bore de stéréochimie Z s'effectue par réaction entre une base faible encombrée comme iPr2NEt (base de Hünig) et un triflate de dialkylebore (R2B-OTf) qui est ici un dérivé du 9-BBN. Du fait de son caractère d'acide de Lewis,
le bore forme un complexe avec l'oxygène du carbonyle tandis que la
base arrache le proton. On obtient ainsi très majoritairement un énolate
de bore de configuration Z ;
Le rapport Z/E > 97 : 3.
- avec des dérivés portant des groupes encombrants sur le bore, comme des groupes cyclohexyle (c-Hex), on peut obtenir l'énolate E quasiment pur comme dans l'exemple suivant ;
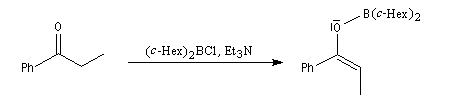
Il y a de très intéressantes informations sur les énolates de bore et leur utilisation en synthèse aux références [69] et [70]. Réaction de Mukaiyama
La réaction de Mukaiyama est un type particulier de condensation aldolique utilisant un éther d'énol silylé. Elle ne procède pas du même mécanisme que l'aldolisation étudiée plus haut. On observe un état de transition ouvert. Réaction de Hajos-Parrish La réaction de Hajos-Parrish est une aldolisation énantiosélective intramoléculaire utilisant comme catalyseur chiral, un aminoacide : la (S)-proline (3 %). On peut la regarder comme un cas particulier d'annélation de Robinson. Dans sa version initiale, le substrat de départ est une tricétone achirale. Le composé obtenu est appelé cétone de Hajos-Parrish.
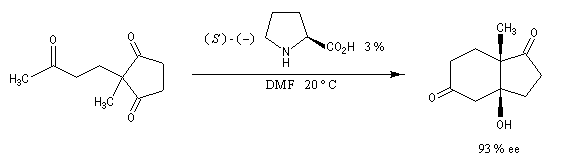
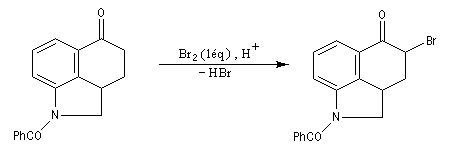
Les composés carbonylés a-halogénés, constituent souvent de très bons substrats pour les réactions de substitution nucléophile SN2 en raison de la mobilité du brome. Dans l'exemple précédent, l'intermédiaire ainsi préparé subit facilement une réaction d'alkylation d'Hofmann permettant l'introduction d'une chaîne latérale sur le cycle.
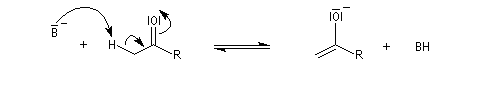
Mécanisme en milieu acide
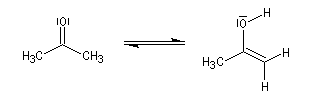
L'étape cinétiquement déterminante est la formation de l'énol.
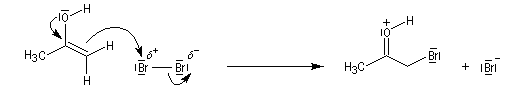
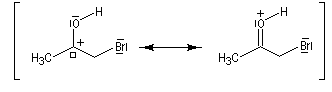


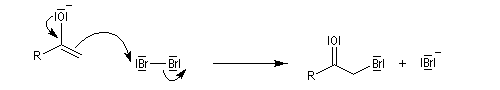
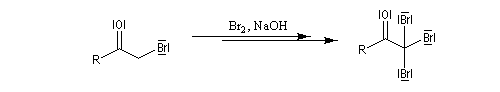

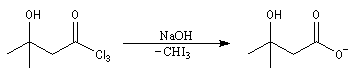
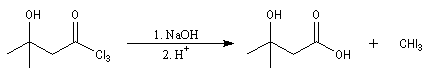
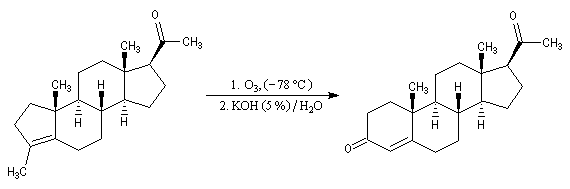
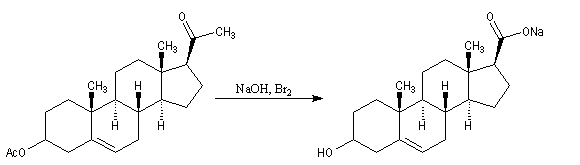
Un autre exemple concerne la préparation d'un dérivé de l'androstérone [63].
 |
L'image de gauche représente un test à l'iodoforme. Compte-tenu de la masse molaire élevée de CHI3 (394 g.mol-1), on commence par dissoudre plusieurs grammes d'iode dans une solution concentrée de soude. Dans ce milieu fortement basique, I2 se dismute en I- et IO3- ce qui se traduit par la décoloration du mélange. On ajoute alors quelques gouttes de composé carbonylé, ici de la propanone. On voit se former rapidement un abondant précipité de CHI3 de couleur blanc-jaunâtre. Cette manière de procéder permet d'éviter que l'iodoforme ne se dissolve dans un excès de composé carbonylé. (La tache brune au fond du tube est due à une petite quantité de diiode non dissous.) |
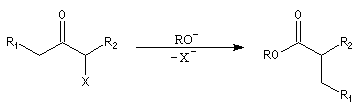
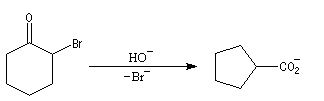
- formation d'un ion énolate ;
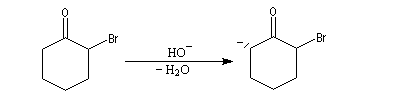
- une alkylation intramoléculaire forme une cyclopropanone ;
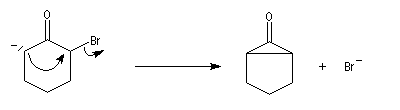
- l'addition d'ions OH- conduit à un intermédiaire tétraédrique instable ;
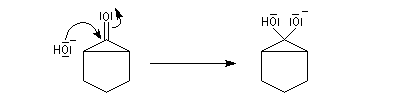
- la cyclopropanone très tendue, est rapidement clivée par la base ;
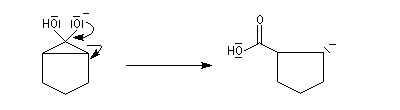
- le carbanion étant beaucoup plus basique que l'ion carboxylate, on assiste à un échange de proton.
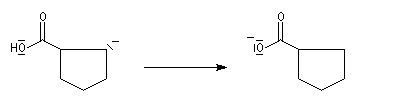
La transposition des a, a'-dibromocétones suivie d'une oxydation puis d'une décarboxylation, est connue sous le nom de réaction de Wallach. Elle permet de passer d'une cycloalcanone à une autre dont le cycle comporte un atome de carbone de moins. Il s'agit donc d'une réaction de rétrohomologation des cétones cycliques.
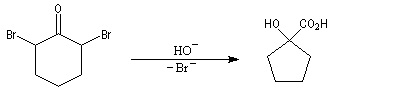
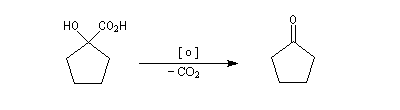
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education, Vol 55, Number 12, December 78.
[12] S. Hünig, E. Lücke, W. Brenninger, Organic Syntheses.
Ouvrages théoriques
[13] J. March - Advanced organic chemistry, Wiley Interscience.
[14] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
[15] N. T. Anh - Introduction à la chimie moléculaire, Ellipses 1994.
[16] J.C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
[17] P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
[19] P. Atkins - General Chemistry, Scientific American Books W. H. Freeman and Co.
[20] J. Koolman, K. H Röhm - Atlas de biochimie, Médecine & Sciences, Flammarion.
[21] A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
Liens
[22] Formaldehyd by W. Wolland - Bellevue Community College, USA
[23] Amygdalin by R. Koepke
[24] H. E. Fischer - The Nobel prize in chemistry 1902
[25] Favorskii
[26] Wittig olefination
[27] Acrolein acetal by J. A. Van Allan
[28] Mc Murry coupling
[29] Wolff-Kishner reduction
[30] Favorskii rearrangement
[31] G. Wittig The Nobel Prize in Chemistry 1979
[32] 2-Naphtoïc acid
[33] Methylencyclohexane by G. Wittig and U. Schoellkopf
[34] Methylencyclohexan oxide by E. J. Corey and M. Chaykovsky
[35] Baeyer-Villiger Oxidation
[36] Assymetric hydrogenation of 3-oxo carboxylate using BINAP-ruthenium complexes by M. Kitamura, M. Tokunaga, T. Ohkuma and R. Noyori
[37] Ethyl cyclohexylideneacetate Horner-Wadsworth-Emmons reaction by W. S. Wadsworth, Jr. and William D. Emmons
[38] Stereoselective Alkene Synthesis : 6-methyl-6-dodecene (Peterson olefination) by Anthony G. M. Barrett, John A. Flygare, Jason M. Hill, and Eli M. Wallace
[39] Wittig-Horner reaction Experimental procedure ; phase transfert catalyst
[50] Strecker synthesis a-aminoisobutyric acid by H. T. Clarke and H. J. Bean
[51] Meerwein-Ponndorf-Verley reduction, trichloroethyl alcohol by Reynold C. Fuson and H. H. Hully
[55] Observation of a Betaïne Lithium Salt during the Course of a Wittig reaction by R. A. Neumann and S. Berger
[57] The Wittig reaction with Pyridylphosphoranes by U. Schröder and S. Berger
[60] Cyclohexylcarbinol by Henry Gilman and W. E. Catlin.
[61] Triphenylcarbinol by W. E. Bachmann and H. P. Hetzner.
[62] Reductive clivage of vinyl phosphorodiamidates by Robert E. Ireland, Thomas H. O'Neil, and Glen L. Tolman
[63] 3-b-acetoxyetienic acid by J. Staunton and E. J. Eisenbraun
[64] International Storehouse Ugo Schiff
[65] Réaction de Hotchkiss-Mac Manus Test APS en cytochimie
[66] Réactif de Feulgen
[67] Carbonyl Chemistry Enolate Formation
[68] From Diyls to Ylides to my Idyll - Nobel Lectures G. Wittig
[69] Enolate chemistry
[70] Z(O,R)-enolates and E(O,R)-enolates
[75] Specific methods in Organic Synthesis
[76] Reductive coupling of carbonyls to alkenes by by Michael P. Fleming and John E. Mc Murry
[77] Tetraphenylcyclopentadienone by John R. Johnson and Oliver Grummitt.
[78] Mistakes, mysteries and errors in chemistry visualization
[90] Réduction de Corey Bakshi Shibata
[90] Cours Crihan IRCOF
Articles
[40] D. Seebach, Synthesis, 1969, 17.
[41] Corey, E. J.; Chaykowsky, M.; J. Am. Chem. Soc., 1965, 87, 1353.
[42] Mc Murry, J. E.; Fleming, M. P.; J. Am. Chem. Soc., 1974, 96, 4708.
[43] H. B. Bürgi, J. D. Dunitz, J. M. Lehn, G. Wipff.; Tetrahedron., 1974, 30, 1563.
[44] Wittig, G.; Geissler, G. Ann., 1953, 580, 44.
[45] L. Horner et al., Ber. 91, 61 (1958); idem et al., ibid. 92, 2499, 1959.
[46] W. S. Wadsworth, Jr., W. D. Emmons, J. Am. Chem. Soc. 1961, 83, 1733.
[47] D. J. Peterson, J. Org. Chem. 33, 780, 1968.
[48] M. Schlosser, K. F. Christmann, Angew. Chem. 1966, 78, 115.
[49] Wittig and Pommer, German Patent 954, 247 (1956), CA 53, 2279, 1959.
[52] H. J. Bestmann, O, Vostrowsky, H. Paulus Tetrahedron Lett., 1977, 121.
[53] J. E. McMurry, J. C. Isser, J. Am. Chem. Soc. 1972, 94, 7132.
[56] E. Vedejs, K. A. J. Snoble, J. Am. Chem. Soc., 1973, 95, 5778-5780.
[58] A. A. Restrepo-Cossio, C. A. Gonzalez, F. Mari, J. Phys. Chem. A. 1998, 102, 6993-7000.
[59] P. J. Kocienski, Tetrahedron Lett., 1979, 2649.
[70] H. E. Zimmerman, M. D. Traxler, J. Am. Chem. Soc. 1957, 79, 1920 1923.
[71] J. Ahman, T. Jarevang, P. Simfai, J. Org. Chem 1996, 61, 8148-8159.
[72] A. E. Arbuzov, J. Russ. Phys. Chem. Soc. , 38, 687 (1906).
[73] R. E. Ireland, J. Am. Chem. Soc. 1976, 98, 2868.
[74] C. Barsu, Correction de l'agrégation externe de chimie 2006, BUP N° spécial 819.
[79] Eisenstein O., Volatron J. Wittig vs Corey-Chaykovsky Reaction. J. Am. Chem. Soc. 1987, vol. 109, n° 1, p 1-14.
[80] Stork, G. ; Cohen, J. F. J. Am. Chem. Soc. 1974, 96, 5270.
[81] R. B. Woodward et al., J. Am. Chem. Soc., 76, 5256 (1954) ;. 78, 3087 (1956)
[82] Asymmetric synthesis of bicyclic intermediates of natural product chemistry Zoltan G. Hajos, David R. Parrish J. Org. Chem. 1974; 39(12); 1615-1621.
[83] V. Prelog, J. Chem. Soc, 1950. 420.
[84] Corey, E. J.; Bakashi, R. K.; Shibata, S. JACS 1987, 109, 7925
[85] T. Ukai, H. Kawazura, Y. Ishii, J. J. Bennett, and J. A. Ibers, J. Organomet. Chem., 65, 253 (1974).
[86] W. R. Bamford and T. S. Stevens J. Chem. Soc., 1952, 4735-4740
[87] A. Wohl, Ber. 26, 730 (1893); 32, 3666 (1899)









0 commentaires:
Enregistrer un commentaire