Cours de chimie Organique -
Composés possédant une liaison azote-azote ou azote-oxygène
Introduction
Classe
Les composés à liaison azote-azote ou azote-oxygène étudiés dans ce chapitre.
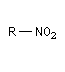 |
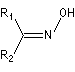 |
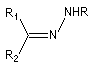 |
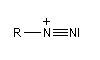 |
|
Dérivé nitré
|
Oxime
|
Hydrazone
|
Ion diazonium
|
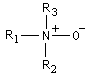 |
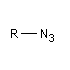 |
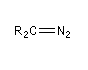 |
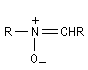 |
|
Oxyde d'amine
|
Azoture
|
Diazoalcane
|
Nitrone
|
Préparation Les dérivés nitrés aliphatiques ou nitroalcanes les plus simples sont obtenus industriellement par nitration d'alcanes en phase vapeur.
Au laboratoire, on peut accéder aux nitroalcanes par substitution à partir d'acides carboxyliques a-halogénés suivie d'une élimination.
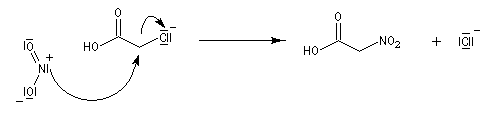
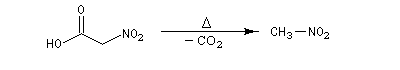
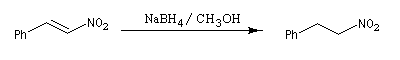 La préparation des dérivés nitrés aromatiques a été étudiée dans le chapitre sur les composés aromatiques.
La préparation des dérivés nitrés aromatiques a été étudiée dans le chapitre sur les composés aromatiques.
Application à la synthèse du nitrostyrène : [18]
Acidité, tautomérie Le groupe nitro est à la fois attracteur d'un point de vue inductif et mésomère.
Les composés qui possèdent un atome d'hydrogène sur le carbone en a du groupe nitro, ont un caractère acide.
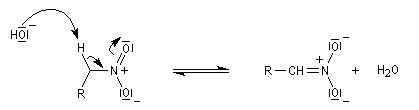
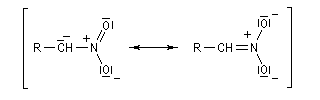
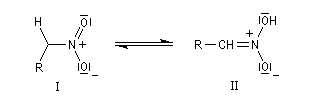
Réduction des dérivés nitrés Les dérivés nitrés aliphatiques peuvent être réduits en amines par simple hydrogénation.
Réaction de Henry Découverte par L. Henry en 1895. Elle est aussi nommée nitro-aldol en raison de sa parenté avec la condensation aldolique.
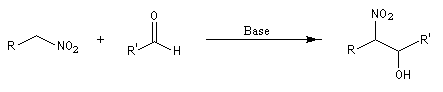
Les composés carbonylés utilisés sont des aldéhydes ou des cétones. Avec ces dernières, la réaction est sensible aux facteurs stériques et conduit généralement à des mélanges de produits dont les proportions dépendent de la nature des réactifs, de la base utilisée, de la température et du temps de réaction.
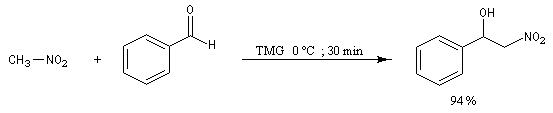
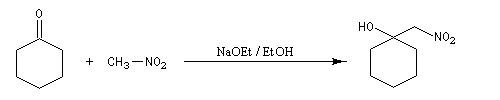
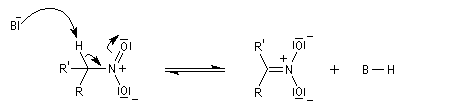
- arrachage d'un atome d'hydrogène sur le carbone en a du groupe nitro avec formation d'un carbanion :
- stabilisation du carbanion par résonance :
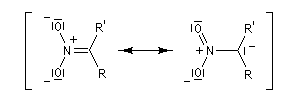
- addition nucléophile du carbanion sur le composé carbonylé ;
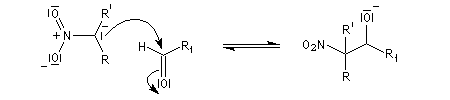
- formation du nitro-alcool et restauration de la base ;
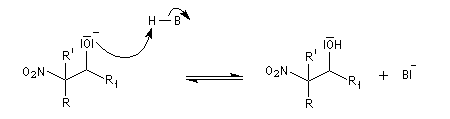
Davantage d'informations sur la réaction de Henry : [32]
La réduction par hydrogénation en présence de Ni de Raney permet de passer des b-nitro-alcools aux b-amino-alcools.
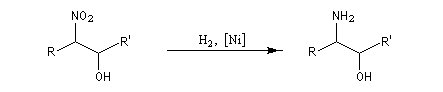
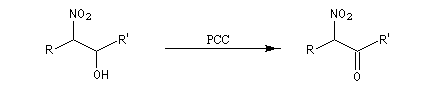
Les carbanions préparés à partir de dérivés nitrés peuvent aussi s'additionnerde façon conjuguée dans une réaction de type Michaël.

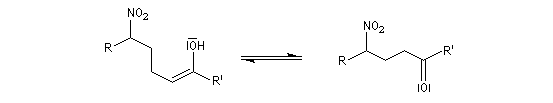
Il y a formation de deux centres chiraux. Il y a donc a priori, formation de quatre stéréoisomères constitués de deux couples d'énantiomères. A cet égard, la situation est assez comparable à celle de la condensation aldol. Cependant, dans les conditions ordinaires, la réaction de Henry n'est pas stéréosélective et on obtient des mélanges de stéréo-isomères. Cela provient du caractère renversable de la réaction et de l'épimérisation facile de l'atome de carbone porteur du groupe nitro.
La première version énantiosélective ("asymétrique") remonte aux travaux du chimiste japonais, Shibasaki en 1992. On l'appelle la Réaction de Shibasaki-Henry. Le catalyseur est un complexe de terre rare utilisant un ligand de type binaphtyle atropique [50]. Cette réaction a été appliquée à la synthèse de la chaîne latérale du taxol.
Davantage d'informations : [40] et [61].
Nitroalcènes
Préparation
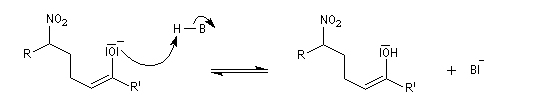
La déshydratation des b-nitro-alcools par un agent déshydratant tel que le dicyclohexylcarbodimide DCC conduit aux nitroalcènes. On obtient un mélange de diastéréo-isomères Z et E .
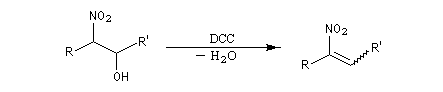
. Propriétés
Les nitroalcènes sont des réactifs utiles dans plusieurs réactions :
- ce sont de bons diénophiles dans la réaction de Diels-Alder en raison du caractère attracteur du groupe nitro ;
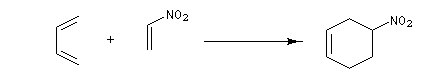
- ce sont des accepteurs dans des réactions de type Michaël.
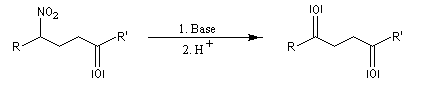
Lorsque l'on traite le sel de sodium du nitroéthane par un acide fort comme H2SO4, on obtient de l'éthanal avec un rendement voisin de 70 %. Cette réaction découverte par le chimiste américain d'origine helvétique John Ulric Nef permet de préparer des composés carbonylés à partir de nitroalcanes.
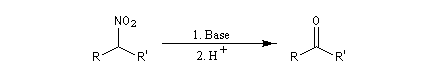
En milieu basique, le dérivé nitré donne facilement un carbanion.
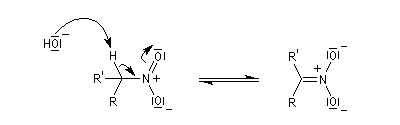
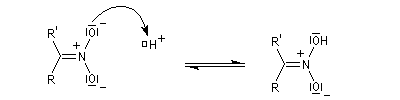
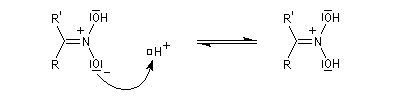
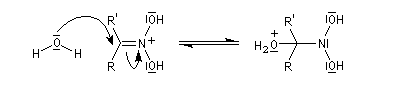
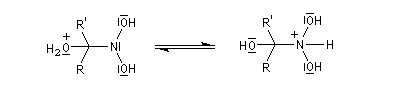
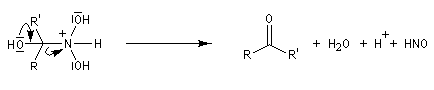
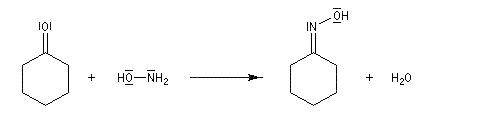
Préparation des oximes Les oximes peuvent être considérées comme le produit de condensation de l'hydroxylamine avec un composé carbonylé.
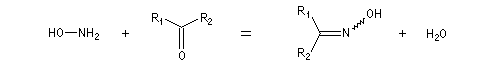
Exemple de préparation : oxime de la benzophénone [19]
Hydrolyse des oximes L'hydrolyse est catalysée par les acides. Elle conduit aux composés carbonylés "parents" aldéhyde ou cétone et à l'hydroxylamine.
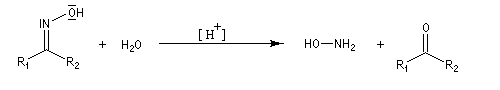
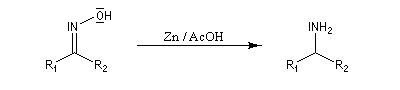
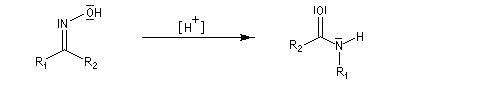
-
protonation ;
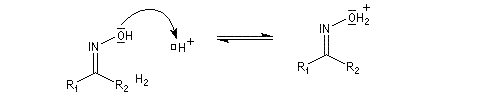
- élimination d'eau ;
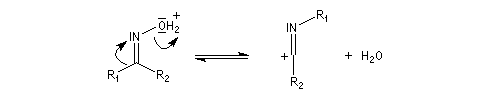
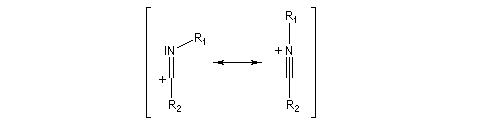
- addition d'eau ;
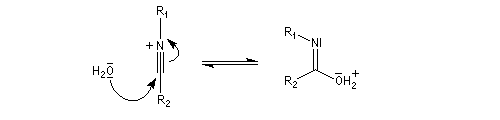
- déprotonation ;
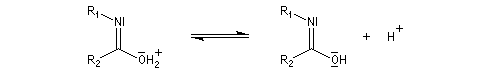
- la dernière étape est une tautomérie iminol-amide.
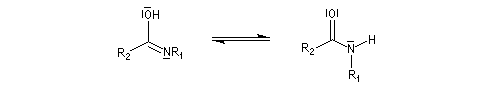
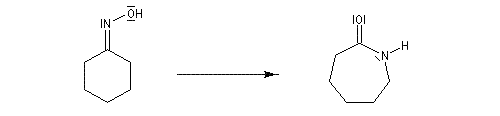
Une illustration importante de la transposition de Beckmann est la préparation du caprolactame.
Dégradation de Wohl
Cette réaction, qui permet de diminuer la chaîne carbonée d'un ose, a été étudiée dans le chapitre consacré aux composés carbonylés.
Hydrazones et dérivés
Préparation L'hydrazine et ses dérivés réagissent avec les composés carbonylés pour donner des hydrazones substituées ou non.
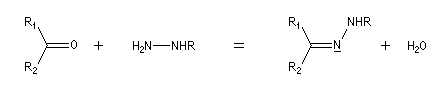
Notons que la réaction de Wolff-Kishner, qui permet la réduction d'un groupe carbonyle en méthylène vis l'hydrazone correspondante a été citée dans ce même paragraphe.
Réaction de Bamford-Stevens En présence de bases très fortes comme des alcoolates, les tosylhydrazones peuvent être converties en composés éthyléniques grâce à la réaction de Bamford-Stevens [46].
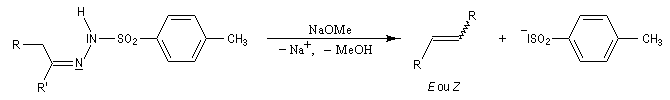
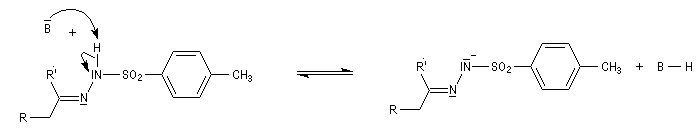
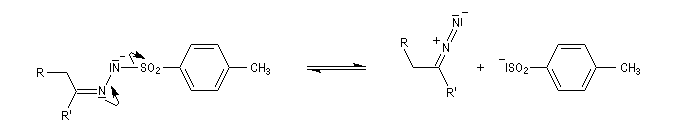
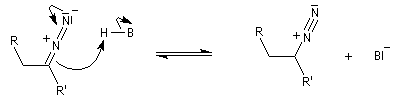
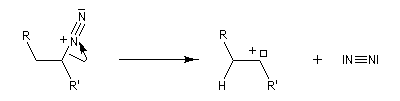
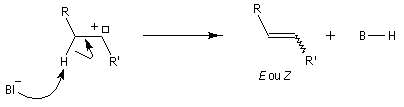
Réaction de Shapiro Il s'agit d'une variante de la réaction de Bamford-Stevens dans laquelle la base forte est un organolithien.
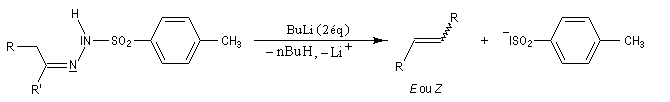
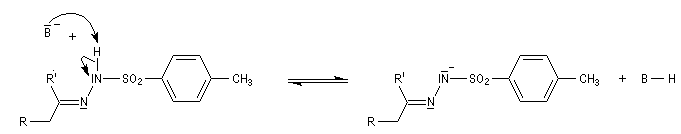
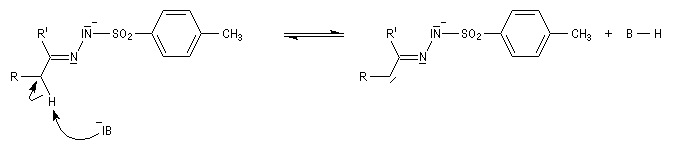
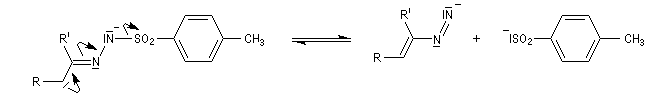
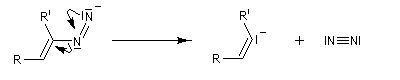
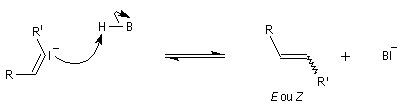
Un exemple d'application à la synthèse du bornène est donné à la référence [39].
La réaction de Shapiro a été utilisée par K. C. Nicolaou dans sa synthèse du Taxol.
Synthèse des indoles de Fischer La découverte de la phénylhydrazine par Fischer en 1875 a largement influencé ses travaux ultérieurs concernant la chimie des sucres et celle de l'indole et de ses dérivés [20].
La réaction entre la phénylhydrazine et un composé carbonylé énolisable, donc possédant au moins un atome d'hydrogène en a du carbonyle, fournit une phénylhydrazone.
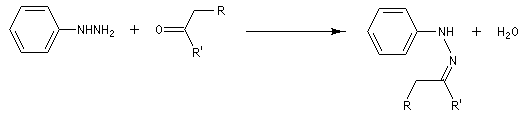
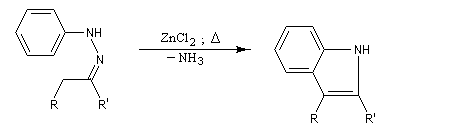
- tautomérie hydrazone-énamine ;
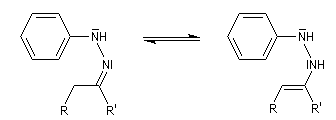
- transposition sigmatropique [3,3] et formation d'une imine (rappel : un autre exemple classique de sigmatropie [3, 3'] est la transposition de Claisen ;
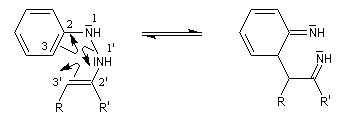
- restauration de l'aromaticité ;
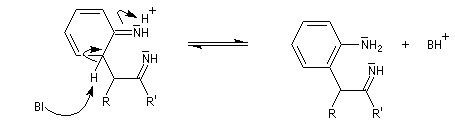
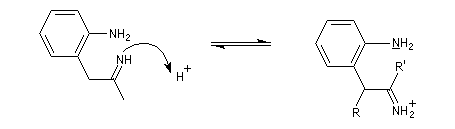
- addition nucléophile sur l'ion iminium qui conduit à une cyclisation ;
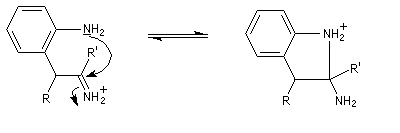
- passage à un aminoacétal (ou aminal : ressemble à un hémiacétal mais un atome d'azote remplace l'atome d'oxygène) ;
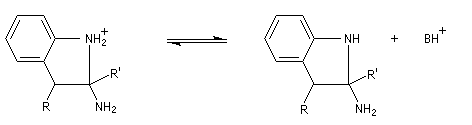
- élimination de NH3 et formation du noyau indole.
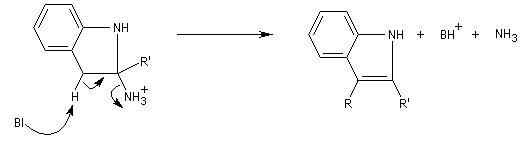
Applications : les dérivés de la triptamine comme le médicament antimigraineux sumatriptan, sont synthétisés selon cette méthode.
De nombreux composés d'origine naturelle contiennent dans leur structure un noyau indole. Citons par exemple l'acide lysergique dérivé de l'ergot de seigle.
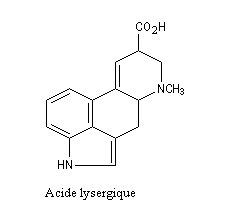 |
L’ergot de Seigle, Claviceps purpurea, est un champignon parasite de certaines céréales (seigle, riz etc) qui contient plusieurs alcaloïdes polycycliques dérivés de l'acide lysergique. Ces composés sont responsables d'une affection appelée ergotisme. La toxine est un puissant vasoconstricteur qui bloque la circulation dans les extrémités. Après une phase convulsive qui peut durer plusieurs semaines, la maladie conduit presque inéluctablement à la mort. Au Moyen Age, cette maladie redoutable était connue sous le nom de "mal des ardents" ou "feu de Saint-Antoine". Information sur les alcaloïdes de l'ergot [17] Synthèse par R. B. Woodward et son équipe [45] |
Préparation des ylures d'ammonium
La découverte des ylures d'ammonium (ou plus simplement : ylure d'azote) remonte aux travaux de G. Wittig. En faisant réagit le méthylithium sur l'ion tétraméthylammonium, on assiste à la métallation d'une liaison C-H du substrat et à la formation d'un ylure d'ammonium (en anglais : ammonium ylides.)
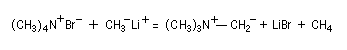
Diazoalcanes
Les diazoalcanes sont des ylures d'alcanediazonium. Leur formule générale peut être représentée par les formes de résonance suivantes :
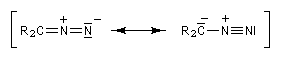
Elles présentent l'intérêt synthétique de conduire aux cétènes via la transposition de Wolff.
Les diazoalcanes non stabilisés sont instables et se décomposent spontanément.
Diazométhane
Le diazométhane est le plus simple des diazoalcanes.
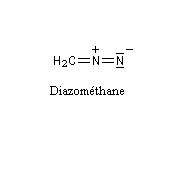 |
Le diazométhane est un gaz de couleur jaune à une température supérieure à - 23 °C (sous 1 bar.) C'est un composé extrêmement toxique, et instable. Sa manipulation (solution à 50 % dans l'éthoxyéthane) nécessite des précautions particulières qui sont rappelées à la référence [15]. Le diazométhane qui n'a pas été utilisé dans une synthèse doit être détruit (il suffit de le mettre en présence d'acide acétique dans l'éther.)
Sa structure est celle d'un ylure d'alcanediazonium.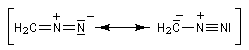 |
- méthylation des acides carboxyliques ;
- méthylation des alcools.
- Préparation des a-diazocétones et réaction d'Arndt-Eistert.
- Addition sur les composés carbonylés.
Préparation des ions diazonium
Les ions diazonium peuvent être préparés par réaction entre une amine primaire et un nitrite en milieu acide. Le réactif électrophile est le cation nitrosonium NO+, formé à partir d'un nitrite en milieu acide.
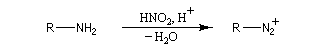
Avec une amine non aromatique, l'ion diazonium est instable et labile. Sa décomposition spontanée fournit un dégagement de diazote, molécule très stable et excellent nucléofuge.
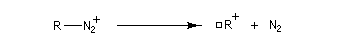
|
butan-1-ol
|
butan-2-ol
|
1-chlorobutane
|
2-chlorobutane
|
|
25 %
|
13 %
|
5 %
|
3 %
|
|
but-1-ène
|
but-2-ène (Z et E)
|
|
26
|
10
|
Application : réaction d'agrandissement de cycle de Demyanov
La réaction de désamination nitreuse peut être mise à profit pour réaliser des agrandissements de cycles grâce à des transpositions de carbocations (réaction de Demyanov).
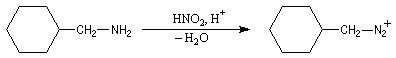
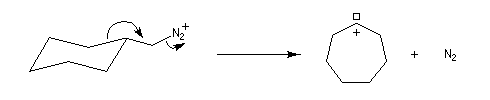
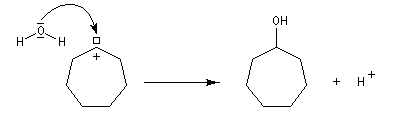
Les solutions d'ions diazonium des amines aromatiques peuvent être conservées plusieurs heures à 0 °C.
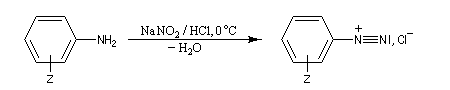
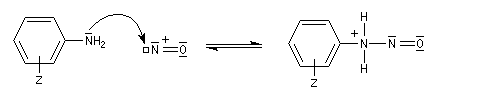
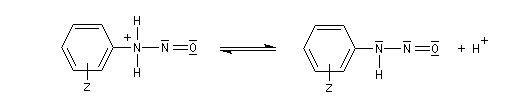
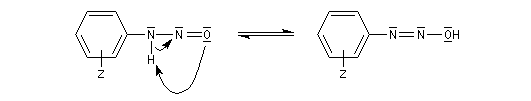
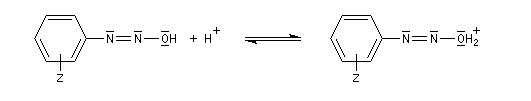
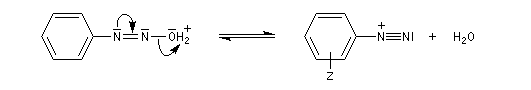
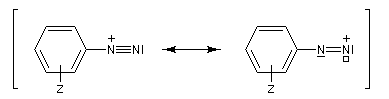
Les ions diazonium réagissent comme des électrophiles assez faibles dans des réaction de substitution aromatique appelées copulations.
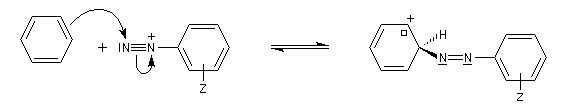
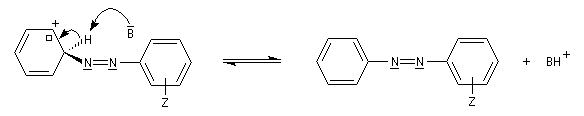
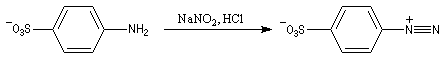
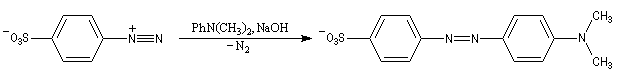
L'hélianthine ou orange de méthyle se présente sous la forme d'un composé solide de couleur orange soluble dans l'eau (I).
 |
 |
 |
|
I
|
II
|
III
|
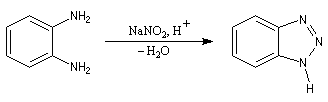
On utilise un réducteur comme EtOH ou H3PO2.
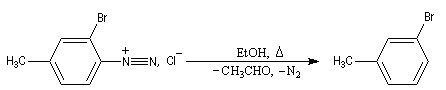
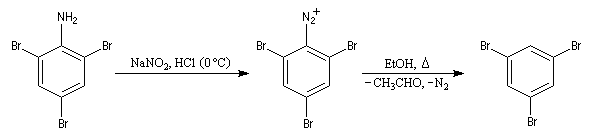
Il s’agit d’une méthode d’iodation des cycles aromatiques. Rappelons que l’iodation d'un cycle aromatique par substitution électrophile n'est pas facilement réalisable(il faut utiliser un mélange de diiode et d'acide nitrique afin de former une petite quantité d'iode cationique.)
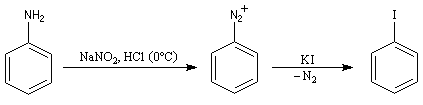
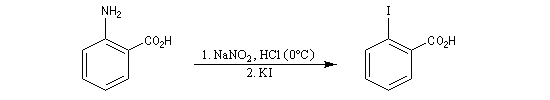
L'iodation d'un cycle aromatique intervient également dans la synthèse de la thyroxine.
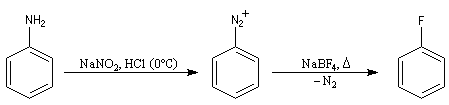
- Dérivés fluorés
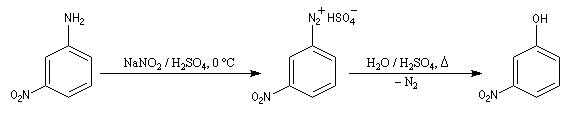
- Passage aux phénols
- Mécanisme
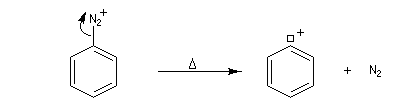
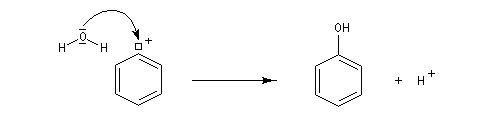
La réaction suit un mécanisme radicalaire. Les réactifs utilisés sont des halogénures de cuivre (I) : CuBr, CuCl. Exemple de protocole : [23]
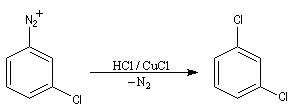
- Dérivés cyanés
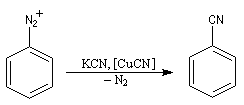
La diazotation de l'acide anthranilique suivie d'une élimination de N2 et de CO2 constitue une méthode douce de préparation du benzyne.
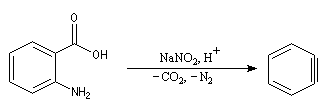
Préparation des oxydes d'amines
Les amines sont des composés assez sensibles à l'oxydation. Les produits de réaction dépendent de la classe de l'amine et de la nature de l'oxydant utilisé. La réaction la plus importante concerne les amines tertiaires. Les oxydants utilisés sont le m-CPBA, le peroxyde d'hydrogène ou encore l'acide peroxymonosulfurique (acide de Caro).
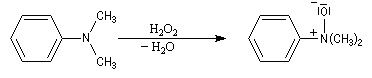
La réaction de Polonovski (Max et Michel Polonovski 1927) met en jeu un oxyde d'amine et un anhydride d'acide comme l'anhydride acétique. Le produit de la réaction est un amide N,N-disubstitué et un aldéhyde (le méthanal si l'oxyde d'amine de départ comporte un substituant méthyle).
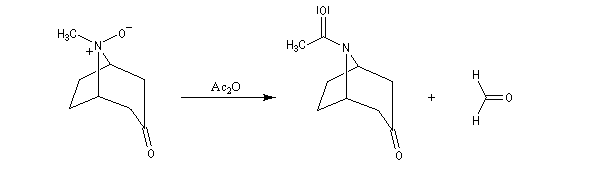
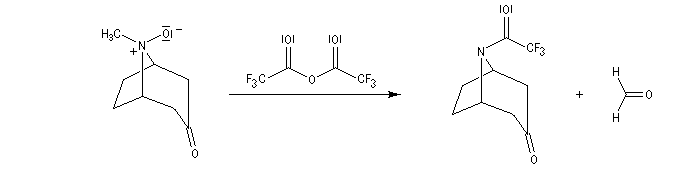
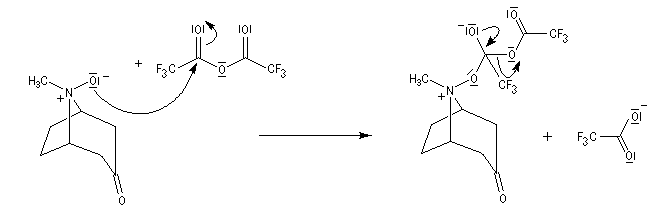
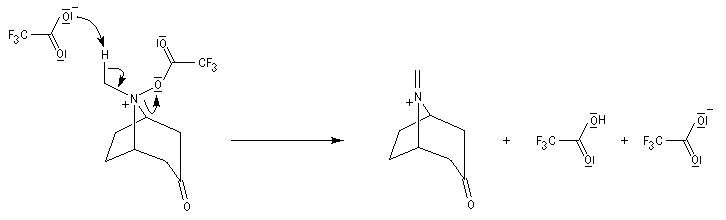
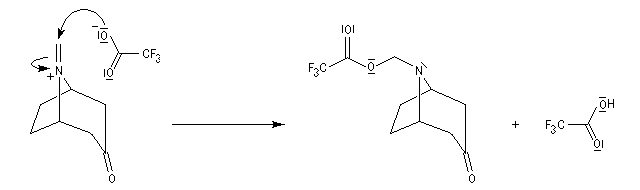
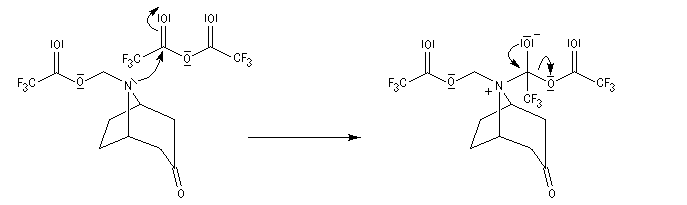
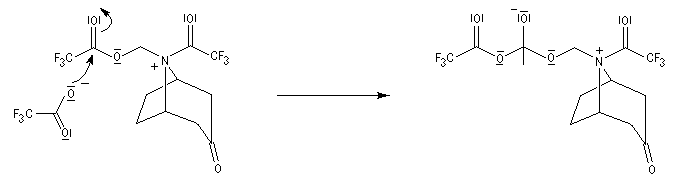
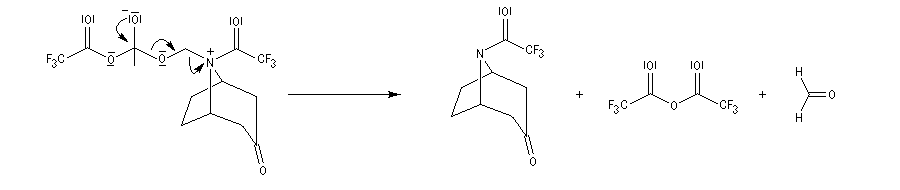
Application de la réaction de Polonovski-Potier à l'hémisynthèse de la vinorelbine
"La chimie est à la biologie ce que le solfège est à la musique" Pierre Potier.
 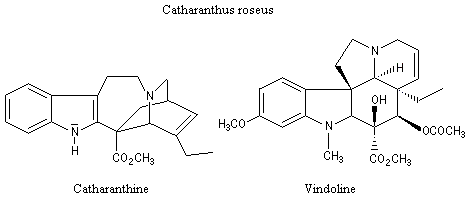
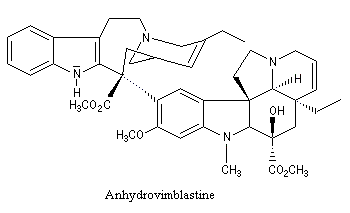
|
La vinorelbine (commercialisée sous le nom de Navelbine®) est un médicament utilisé en chimiothérapie anticancéreuse. Sa synthèse s'effectue à partir de deux molécules extraites de la pervenche de Madagascar (Canthareus roseus) : la catharanthine et la vindoline. Le couplage chimique de ces sous-unités conduit dans un premier temps à l'anhydrovimblastine qui est ensuite transformée en vinorelbine par une suite de réactions dont l'étape clé est la réaction de Polonovski-Potier. Il faut extraire environ 20 tonnes de feuilles séchées pour produire 1 kilogramme du médicament [42]. |
Réaction entre l'oxyde d'amine et l'anhydride d'acide.
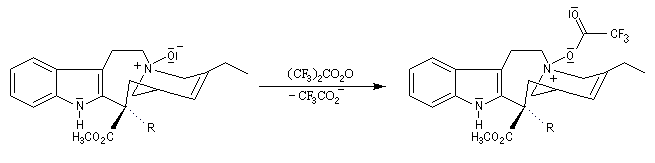
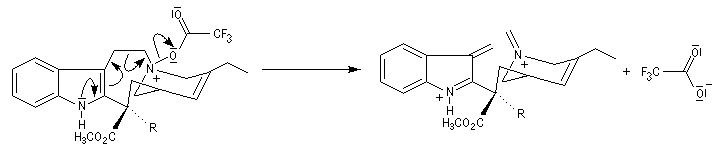
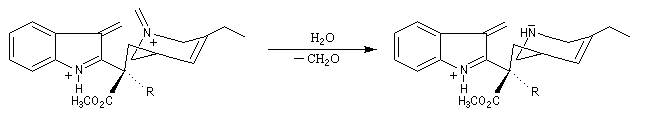
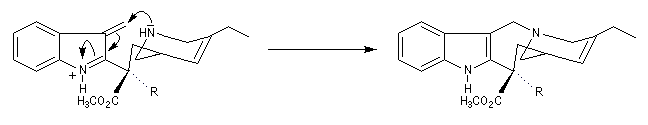
Elimination de Cope des oxydes d'amines
L'une des réactions importante des oxydes d'amines est l'élimination de Cope qui conduit aux composés éthyléniques. Il s'agit d'une b-élimination. L'atome d'oxygène de l'oxyde joue le rôle de base tandis que l'atome d'azote positif est le nucléofuge. La réaction possède l'avantage d'être stéréospécifique de stéréochimie syn. L'exemple suivant concerne la préparation du méthylènecyclohexane [24].
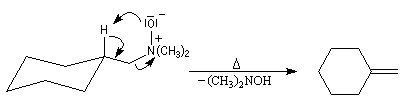
Article original : [44]
Azotures d'alkyles
Préparation
On peut préparer les azotures d'alkyle par une réaction de substitution nucléophile sur un dérivé halogéné comme substrat. Le nucléophile est l'ion azoture (en anglais : azide) dont une source commerciale est l'azoture de sodium. Ce réactif est à la fois explosif et toxique .
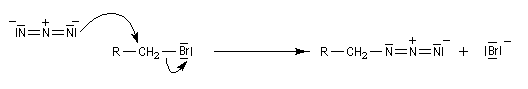
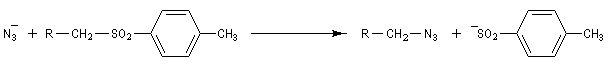
Les azotures d'alkyles sont facilement réduits en amines par hydrogénation catalytique ou en utilisant LiAlH4. Puisque les dérivés halogénés et les dérivés sulfonylés d'alcanes sont aisèment obtenus à partir des alcools, les azotures d'alkyles permettent le passage d'un alcool à une amine.
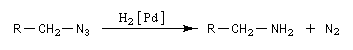
Réaction de Staudinger (aza-Wittig)
Réaction mise au point par H. Staudinger [49] La triphénylphosphine réagit sur un azoture pour donner un iminophosphorane.
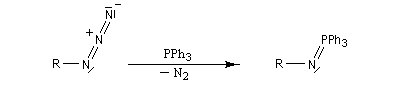
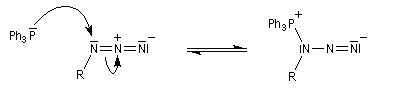
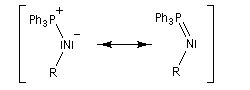
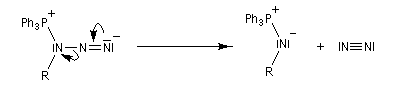
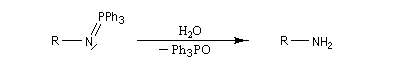
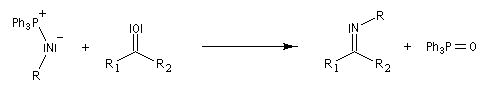
Transposition de Curtius
L'ion azoture réagit avec les chlorures d'acycles pour donner des acylazides.
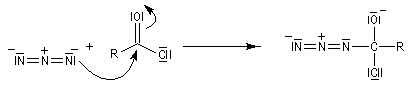
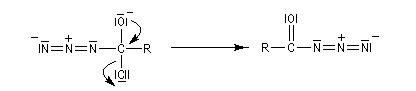
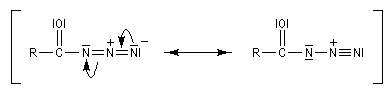
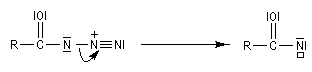
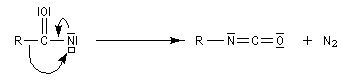
En présence de tBuOH, on obtient des amines protégées par le groupe Boc (t-butoxycarbonyle).
Voir également :
- transposition de Wolff ;
- réaction de Schmidt découverte par Karl Friedrich Schmidt en 1924 [27].
Préparation des nitrones
Les nitrones sont des N-oxydes d'imines. On peut les obtenir par plusieurs méthodes :
- oxydation d'hydroxylamines N,N disubstituées [36].
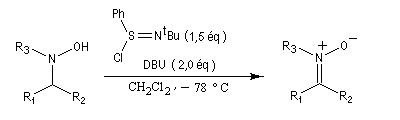
- condensation d'une hydroxylamine monosubstituée avec un composé carbonylé.
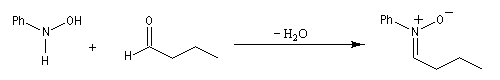
Cycloaddition [3 + 2]
L'une des réactions les plus importantes des nitrones est leur cycloaddition avec les alcènes ou les alcynes [60].
La réaction entre une nitrone (dipôle) et le composé éthylénique (dipolarophile) peut conduire a priori à plusieurs composés. La régiochimie de l'addition peut être bien rationalisée en prenant en compte l'interaction entre les orbitales frontières des réactifs.
[38] Préparation de nitrones et cycloaddition-1,3-dipolaire.
Bibliographie
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education vol 55, 1977.
[12] S. Hünig, E. Lücke, W. Brenninger, Organic Syntheses, V, p 533.
Liens
[13] Nitronates by Scott E. Denmark and Jeromy J. Cottell
[14] Préparation of Nitromethane by F. C. Whitmore and Marion G. Whitmore
[15] Diazomethan by Th. J. de Boer and H. J. Backer
[16] Montage de distillation pour la préparaton du diazométhane
[17] Le mal des ardents : la chimie de l'ergot de seigle
[18] Nitrostyrène by David E. Worrall
[19] Benzophenone oxime Arthur Lachman
[20] Emil Fischer, prix Nobel de chimie 1902
[21] R. B. Woodward - The Nobel Prize in chemistry 1965
[22] p-fluorobenzoic acid by G. Schiemann and W. Winkelmüller
[23] 1-chloro-2,6-dinitrobenzene Organic Syntheses, Vol. 32, p. 23 (1952); Coll. Vol. 4, p.160 (1963)
[24] Preparation of methylenecyclohexane by Arthur C. Cope and Engelbert Ciganek, Massachusetts Institute of Technology, Cambridge 39, Massachusetts
[25] m-nitrobenzazid Organic Syntheses, Vol. 33, p. 53 (1953); Coll. Vol. 4, p.715 (1963).
[26] 2-phenyl-2-adamentanamine hydrochloride Organic Syntheses, Vol. 60, p. 104 (1981) ; Coll. Vol. 7, p.433 (1990).
[27] Highlights of Shmidt reaction in the last ten years
[28] The synthesis of laurolactame from cyclodecanone via a Beckmann rearrangment
[29] 1-Morpholino-1-cyclohexene by Hünig, E. Lücke, and W. Brenninger
[30] Hofmann rearrangement under mildy acidic condition by Merrick R. Almond, Julie B. Stimmel, E. Alan Thompson, and G. Marc Loud
[31] carbon dehomologation reactions : Schmidt, Curtius, Hofmann, Hunsdiecker by R. M. Williams
[32] Henry reaction
[33] Assymetric Henry and aza Henry reaction by D. J. Osborn
[34] Réarrangement du squelette de la catharanthine
[35] Nitrone-olefin [3+2] cycloadditions
[36] A convenient method for synthesis of nitrone by Teruaki Mukaiyama
[37] Pierre Potier, médaille d'or du CNRS
[38] Nitrones for 1,3-dipolarcycloadditions by Norman A. LeBel and Dorothy Hwang.
[39] 2-bornène by R. H. Shapiro and J. H. Duncan.
[60] 1,3-dipolar Cycloaddition Reaction of Nitrones with Alkenes.
[61] Asymetric variants of the Henry reaction by J. Wilmot.
Ouvrages théoriques
A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
J. March - Advanced organic chemistry, Wiley Interscience
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
P. Atkins - General Chemistry, Scientific American Books W. H. Freeman and Co.
J. Koolman, K. H Röhm - Atlas de biochimie, Médecine & Sciences, Flammarion.
P. Caubère - La catalyse par transfert de phase et son utilisation en chimie organique, Masson 1982.
Organic Synthesis Highlights V, Edited by Hans-Günther Schmalz and Thomas Wirth, J. Wiley 2003.
Articles
[40] M. Shibasaki, H. Sasay, T. Aray, Angew. Chem. Int. Ed. Engl., 36, 1236, (1997)
[41] G. Stork, Tetrahedron, 1982, 38, 1975, 3363
[42] Produits naturels anticancéreux, Daniel Guénard, Françoise Guéritte, Pierre Potier, l'actualité chimique, avril-mai 2003.
[43] Robinson, R. J. Chem. Soc. 1917, 111, 762-76 .
[44] Thermal Decomposition of Amine Oxides to Olefins and Dialkylhydroxylamines, Arthur C. Cope, Theodore T. Foster, Philip H. Towle J. Am. Chem. Soc., 1949, 71 (12), pp 3929–3934
[45] Edmund C. Kornfeld, E.J. Fornefeld, G. Bruce Kline, Marjorie J. Mann, Dwight E. Morrison, Reuben G. Jones and R.B. Woodward
[46] W. R. Bamford and T. S. Stevens J. Chem. Soc., 1952, 4735-4740.
[47] Shapiro, R.H.; Lipton, M.F.; Kolonko, K.J.; Buswell, R.L.; Capuano, L.A. Tetrahedron Lett., 1975, 1811
[48] Fischer. E. ; Jourdan, F. Chemische Berichte 1883, 16, 2241
[49] Staudinger, H.; Meyer, J. Hel. Chim. Acta 1919, 2, 635
[50] Sasai, H.; Takeyuki, S.; Arai, S.; Arai, T.; Shiba, J. Am. Chem. Soc, 1992, 114, 4418-4420.









0 commentaires:
Enregistrer un commentaire