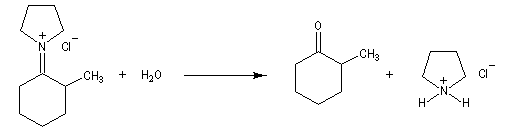
Cours de chimie Organique -
Amines et composés apparentés
Introduction
Classe
Les amines sont des composés azotés qui dérivent formellement de l'ammoniac NH3 par remplacement d'un ou plusieurs atomes d'hydrogène par des groupes carbonés. Le nombre n des atomes d’hydrogène liés à l'azote, définit la classe de l’amine. Leur découverte est due au chimiste allemand Wurtz en 1849.
|
n
|
2
|
1
|
0
|
|
Classe
|
primaire
|
secondaire
|
tertiaire
|
- Acyclique : l'atome d'azote est relié à un ou plusieurs groupes alkyles. Exemple : la méthylamine CH3NH2.
- Alicyclique : l'atome d'azote est lié à un cycle non aromatique. Exemple : la cyclohexylamine.

- Aromatique : l'atome d'azote est lié à un cycle aromatique. Exemple : la phénylamine ou aniline.
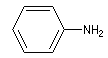
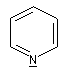
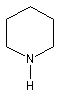
- Hétérocyclique : l'atome d'azote est engagé dans un cycle qui peut être ou non aromatique. Exemples :
 |
 |
|
pyridine
|
pipéridine
|
Les amines acycliques forment des série homologues appartenant aux trois classes. Les amines et des diamines simples qui sont assez volatiles possèdent une odeur souvent nauséabonde. Celle de la triméthylamine rappelle le poisson pourri. La pentane-1,5-diamine qui se forme lors de la décomposition des protéines s'appelle vulgairement cadavérine.
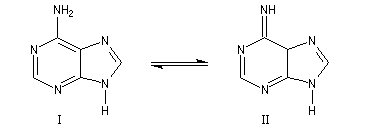
Bases puriques et pyrimidiques
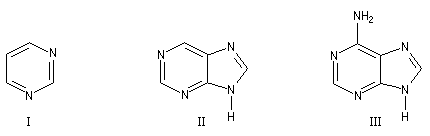
Les bases pyrimidiques et puriques, dérivées respectivement de la pyrimidine (I), de la purine (II), sont des constituants des acides nucléiques. L'adénine (III), intervient dans la structure de l'ATP.
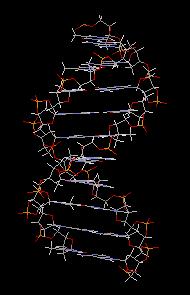 |
James Watson, Francis Crick ont proposé la structure
en double hélice de l'ADN dans un célèbre article de la revue Nature en
1953 [47].
Leur argumentation s'appuyait notamment sur les travaux de diffraction
aux rayons X de Maurice H. F. Wilkins et Rosalind Elsie Franklin (King's
College
, Londres.) [39]. Watson, Crick et Wilkins ont obtenu le prix Nobel de physiologie et médecine en 1962. [30]. Compléments sur les propriétés physico-chimiques de l'ADN [22]. (photographie G. D. 2011.)
 |
Les molécules hétérocycliques, azotées, d'origine naturelle s'appellent alcaloïdes en raison de leurs propriétés basiques. Ils possèdent généralement une activité biologique marquée. Donnons quelques exemples d'alcaloïdes.
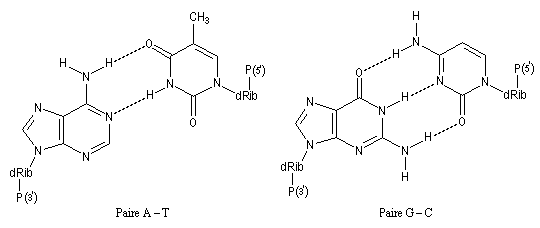
- La caféine (C) peut être extraite du café et du thé [2] et [5]. La théobromine (T) est contenue dans le cacao.
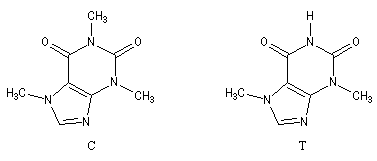
- La quinine,
présente dans l'écorce de quinquina, a été le premier médicament
réellement efficace contre le paludisme. Malheureusement la toxicité du
composé limite son champ thérapeutique. On le réserve dans le traitement
de certaines formes aigües qui résistent aux autres antimalariques.

La découverte des vertus thérapeutiques des dérivés du quinquina remonte au 17ème siècle. La légende veut que la comtesse de Chinchona, femme d'un Roi du Pérou, ait été guérie d'une fièvre (due à la malaria ?) grâce à des décoctions d'écorce de cet arbre. Le principe actif a été isolé par deux pharmaciens français en 1820, Pierre-Joseph Pelletier et Joseph-Bienaimé Caventou. En 1908, P. Rabe établit la formule brute correcte de la quinine : C20H24N2O2. En 1856, l'impétueux William Perkin, âgé de seulement 18 ans, tente de synthétiser la quinine afin de soigner les combattants britanniques victimes de la malaria en Inde. Se fondant sur la formule brute de la quinine, il recherche des substrats susceptibles de l'engendrer. Considérant l'allyltoluidine comme un bon candidat, il envisage son oxydation selon : 2 C10H13N + O2 = C20H24N2O2 + H2O
Inutile de dire qu'il ne parvient pas à ses fins. Mais cet échec le conduit à la découverte du premier colorant synthétique : la mauvéine. Il fonde une entreprise afin de commercialiser sa découverte. C'est le début de l'industrie des colorants [19].
La première synthèse stéréosélective de la quinine a été publiée en 2001 par Gilbert Stork (Columbia University). La molécule possède quatre centres chiraux. Par exemple : la quinine et la quinidine sont des diastéréo-isomères qui diffèrent par la configuration du centre chiral porteur du groupe OH. Notons que la quinine fait partie des molécules naturelles du fonds chiral (chiral pool en anglais). A ce titre, elle est utilisée, ainsi que ses dérivés, comme auxilliaire chiral dans plusieurs réactions énantiosélectives. La plus célèbre étant la dihydroxylation asymétrique de Sharpless.
Plusieurs synthèses totales de la quinine ont été publiées : Stork (2001), Jacobsen (2004), Kobayashi (2004). Les grandes lignes de ces synthèses sont données à la référence : [35].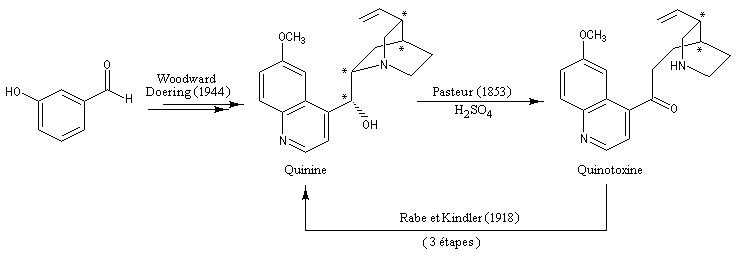
Remarque : la quinine possède la propriété d'être fluorescente sous l'action d'un rayonnement UV de longueur d'onde voisine de 350 nm. La fluorescence s'effectue dans le visible aux alentours de 450 nm. Cette propriété permet son dosage [44].
Le professeur R. B. Woodward (Université de Harvard) est considéré comme l'un des chimistes organiciens les plus brillants du 20 ème siècle. Le prix Nobel de chimie lui a été attribué en 1965 pour l'ensemble de ses travaux de synthèse organique et notamment la synthèse totale de nombreux composés complexes d'origine naturelle. Parmi les nombreuses anecdotes qui émaillent la carrière de ce chimiste hors du commun, signalons simplement qu'il conçut les grandes lignes de la synthèse de la quinine alors qu'il était encore jeune élève au lycée. R. B. Woodward est décédé en 1979 [21], [25].
- Certains alcaloïdes sont des poisons violents. La strychnine et la brucine sont présentes à l'état naturel dans la noix vomique. Elles ont été utilisées comme poison de flèche. L' acide lysergique et ses dérivés sont des composés très toxiques produits par l'ergot de seigle. La coniine est un dérivé de la pipéridine.
 |
La coniine est la 2-propylpipéridine. A. Ladenburg a séparé les deux énantiomères en 1886. La (S)-(-)-coniine est l'un des alcaloïdes
de Cicuta naculata une ombellifère plus connue sous le nom de cigüe.
On la trouve dans la nature le long des chemins. Ce poison était
utilisé par
les grecs pour exécuter les condamnés à mort. C'est après avoir absorbé
une solution de cigüe que le philosophe Socrate, mourut en - 399 [17]. |
- La strychnine et la brucine sont des amines tertiaires chirales. Elles peuvent être obtenues assez facilement sous forme énantiopure. C'est la raison pour laquelle ont les utilise comme agents résolvants ou comme auxiliaire chiral dans certaines synthèses asymétriques.
- La dihydroquinine (DHQ) et la dihydroquinidine (DHQD) entrent dans la composition de catalyseurs asymétriques permettant la dihydroxylation énantiosélective des doubles liaisons éthyléniques, réaction mise au point par K. B. Sharpless en 1987.
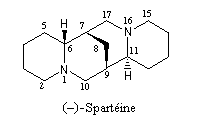
- La spartéine, un alcaloïde présent à l'état naturel dans le lupin blanc, constitue un ligand chiral dans les réactions de déprotonation énantiosélectives par des organolithiens.
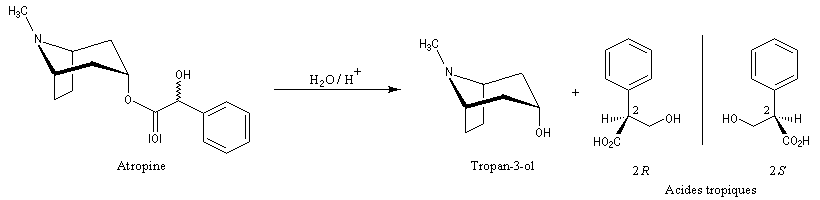
- Par hydrolyse des alcaloïdes, on obtient souvent des molécules plus simples. L'hydrolyse de l'atropine, composé existant dans certaines plantes de la famille des solanacées fournit le tropan-3-ol, achiral (composé méso) et le mélange racémique des acides tropiques (3-hydroxy-2-phénylpropanoïques.)
- De nombreuses toxines possèdent dans leur molécule une ou plusieurs fonctions amine. La tétrodotoxine est présente à l'état naturel chez plusieurs animaux et notamment dans le poisson tétrodon. Les cuisiniers japonais doivent être titulaire d'un diplôme spécial pour préparer ce plat très apprécié au pays du Soleil levant mais qui provoque plusieurs intoxications mortelles chaque année. C'est en effet l'un des poisons les plus violents connus. Sa structure a été déterminée conjointement par R. B. Woodward et T. Kishi en 1965. Une monographie consacrée à cette molécule se trouve à la référence [15].
- La morphine est l'un des antalgiques les plus puissants connus.
Méthylamine
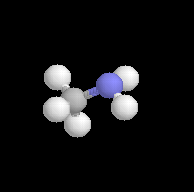 |
La méthylamine est préparée par alkylation de l'ammoniac. |
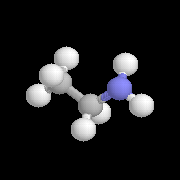 |
L'éthylamine est préparée industriellement par amination réductive de l'éthanal. . |
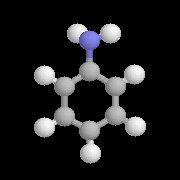 |
C'est l'amine aromatique la plus importante. On l'obtient par réduction du nitrobenzène [6] par un métal en
milieu acide.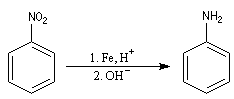 |
La méthode qui consiste à considérer une amine comme un dérivé substitué du nitrure d'hydrogène correspondant, appelé azane (l'ammoniac NH3 est l'azane le plus simple), n'est presque jamais employée [40].
Nomenclature radico-fonctionnelle
On ajoute la terminaison amine au nom du groupe lié à l'atome d'azote. L'atome de carbone lié à l'azote porte le numéro 1.
|
|
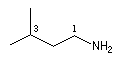 |
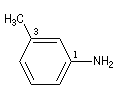 |
|
propylamine
|
3-méthyl-1-butylamine
|
3-méthyl-1-phénylamine
|
- Amines secondaires et tertiaires
 |
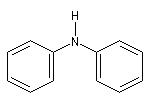 |
|
diéthylamine
|
diphénylamine
|
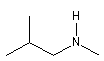 |
 |
|
N-méthyl-2-méthylpropylamine
|
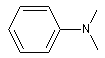
N, N-diméthylphénylamine
|
- Amines hétérocycliques
 |
 |
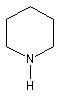
|
azacyclohexane
|
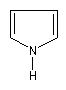
1-azacyclopenta-2,4-diène
|
Nomenclature substitutive
On ajoute la terminaison amine au nom de l'alcane après avoir supprimé la lettre e.
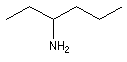 |
|
|
hexan-3-amine
|
hexane-1,6-diamine
|
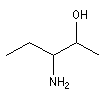 |
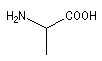 |
|
3-aminopentan-2-ol
|
acide 2-aminopropanoïque
|
Propriétés physiques
Température de changement d'état
Les propriétés physiques sont influencées par l'existence de liaisons hydrogène qui possèdent toutefois une intensité moindre que chez les alcools de masse molaire comparable car l'atome d'azote est moins électronégatif que l'atome d'oxygène. Les amines de faible masse molaire sont miscibles à l'eau.
|
Composé
|
M (g.mol-1)
|
TE (°C)
|
m (D)
|
(20 °C, 1 bar)
|
|
CH3CH2OH
|
46
|
78,5
|
1,71
|
liq
|
|
CH3CH2NH2
|
45
|
16,6
|
1,29
|
gaz
|
L'atome d'azote est au sommet d'une pyramide y compris dans l'aniline ce qui est conforme aux prévisions de la méthode VSEPR. L'angle a entre les liaisons est tel que a < 109° du fait de la présence du doublet non liant. Dans la plupart des cas, la structure n'est pas rigide. La barrière énergétique séparant les configurations est généralement faible et la fréquence d'interconversion est très élevée.
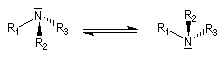
|
E (kJ.mol-1)
|
n (MHz)
|
|
30
|
103
|
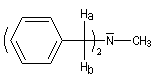
L'inversion peut quelquefois être assez lente pour que les deux énantiomères puissent être identifiés, voire piégés, à condition d'opérer à une température suffisamment basse. On rencontre ce type de situation dans la famille des aziridines substituées à l'azote.
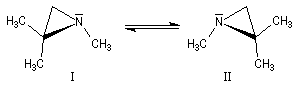
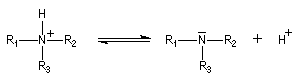
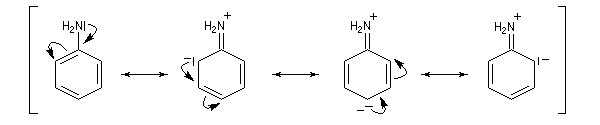
- Amines aromatiques
|
ER (kJ.mol-1)
|
m (D)
|
|
172
|
1,56
|
Spectroscopie infrarouge
La spectroscopie infrarouge constitue une méthode de choix pour l'identification des structures azotées. Les amines primaires et secondaires peuvent en général être distinguées car elles possèdent des spectres assez caractéristiques.
|
s (cm-1)
|
3500 - 3400
|
1650 - 1550
|
1350 - 1250
|
|
Vibration
|
élongation N-H (doublet)
|
déformation N-H
|
élongation C-N
|
s (O-H) = 3600 cm-1.
La fréquence d'absorption de la liaison C-N de l'aniline apparaît vers s (C-N) = 1300 cm-1 ce qui traduit un renforcement de la liaison dû à la participation à la résonance du doublet de l'azote.
On observe un doublet chez les amines primaires qui est dû au couplage entre les deux vibrateurs N-H. Les vibrations peuvent être symétriques ou non symétriques.
- Amines secondaires
|
s (cm-1)
|
3400-3300
|
1600-1490
|
1350-1250
|
|
Vibration
|
élongation N-H (simple)
|
déformation N-H
|
élongation C-N
|
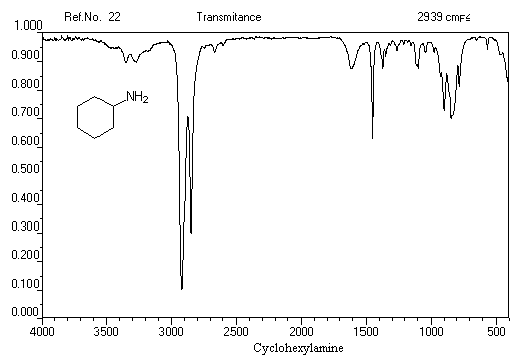
Exemple 1 : spectre IR de la cyclohexylamine
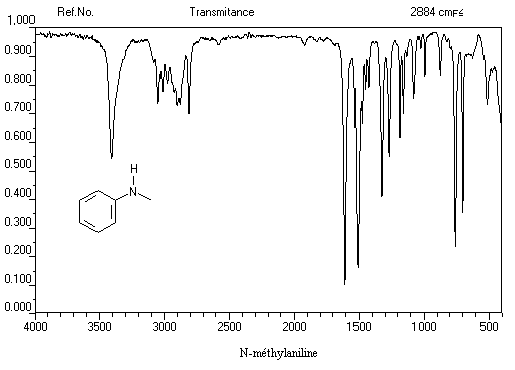
Exemple 2 : spectre IR de la N-méthylaniline
Voici un tableau présentant les déplacements chimiques les plus importants du proton lié à l'azote. On a fait figurer les amides dans le tableau à titre de comparaison.
|
d (ppm)
|
0 - 2
|
2 - 5
|
5 - 9
|
|
Composé
|
amines non aromatiques
|
amines aromatiques
|
amides
|
Un moyen d'identifier les protons liés à l'azote consiste à enregistrer le spectre dans un solvant inerte puis en présence d'acide trifluoroacétique qui protone l'amine. Le pic du proton lié à l'azote possède un déplacement chimique plus grand dans le cation que dans l'amine.
La spectroscopie dans l'ultraviolet et le visible est surtout utile pour les amines aromatiques. Le spectre ci-dessous est celui de l'aniline.
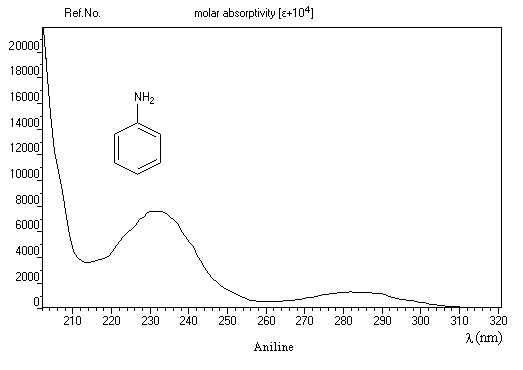
|
Composé
|
bande E
|
bande B
|
|
benzène
|
203 nm (e = 7400)
|
254 nm (e = 204)
|
|
aniline
|
235 nm (e = 8600)
|
285 nm (e = 1430)
|
|
ion anilinium
|
203 nm (e = 7500)
|
254 nm (e = 169)
|
Lorsque la conjugaison s'étend sur un plus grand nombre d'atomes comme dans les composés diazoïques, la longueur d'onde du maximum d'absorption peut se trouver dans le domaine visible. Le composé est coloré. L'hélianthine est un composé dont la couleur dépend de l'extension du système conjugué avec le pH.
Propriétés acido-basiques
Basicité
Elle est due au doublet non liant porté par l'atome d'azote. La plupart des amines manifestent des propriétés basiques en solution aqueuse. La réaction prépondérante avec l'eau peu s'écrire :
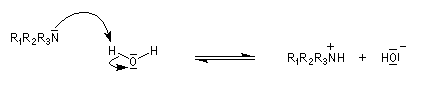

|
Composé
|
PhNH2
|
PyH
|
NH3
|
Me3N
|
MeNH2
|
Me2NH
|
|
pKa
|
4,6
|
5,6
|
9,2
|
9,8
|
10,6
|
10,7
|
secondaires > primaires > tertiaires
Cet ordre n'est pas facile à rationaliser car les écarts observés ont
une faible amplitude. Plusieurs effets possédant le même ordre de
grandeur se superposent : électroniques, stériques, et surtout
solvatation.En série cyclique, la situation est plus compliquée. Ainsi, l'aniline est 100 000 fois moins basique que la cyclohexylamine : pKa (CyNH3+/CyNH2) = 9,6 ; pKa (PhNH3+/PhNH2) = 4,6. On interprète habituellement ce résultat par le fait que le doublet non liant de l'aniline est moins disponible dans cette amine aromatique que dans la cyclohexylamine du fait de la délocalisation électronique dans le premier cas. Cependant, cet effet n'est pas isolé ; la solvatation joue aussi un rôle important.
Le cas des amines hétérocycliques aromatiques est aussi un sujet délicat.
- Dans la pyridine, le doublet de l'azote n'est pas impliqué dans le système aromatique. La pyridine est basique.
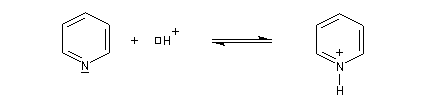
- En revanche, dans le pyrrole, le doublet participe à la
conjugaison au sein d'un cycle aromatique. Le pyrrole ne manifeste pas
de propriétés basiques.
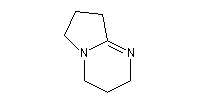
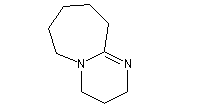
- DBN et DBU sont utilisées comme base dans de nombreuses réactions comme par exemple la réaction de Wadworth-Emmons ou encore la réaction de Henry.
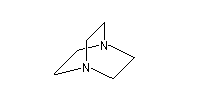
- DABCO et DBU sont utilisées comme catalyseur dans la réaction de Baylis-Hillman.
 |
 |
 |
|
1,5-diazabicyclo[4.3.0]non-5-ène
(DBN)
|
1,8-diazabicyclo[5.4.0]undec-7-ène
(DBU)
|
1,4-diazabicyclo[2.2.2]octane
(DABCO)
|
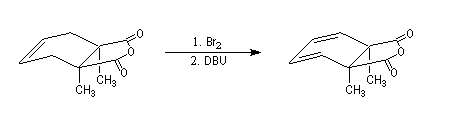
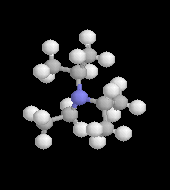 |
L'image de gauche représente la molécule de N,
N-diisopropyléthylamine en core appelée base de Hunig. Elle peut
remplacer la triéthylamine comme capteur de protons dans de nombreuses
applications. Elle présente en outre plusieurs avantages par rapport à
cette dernière :
|
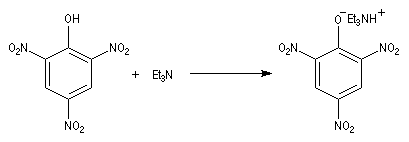
La basicité des amines est mise à profit pour leur extraction d'un milieu réactionnel.
- en milieu aqueux, en présence d'acides forts, on obtient des sels d'aminium solubles dans l'eau ;
- il est possible d'opérer en milieu non aqueux avec des solutions de HCl dissous dans l'éther. Dans ce cas, le chlorhydrate d'aminium précipite en milieu inorganique.
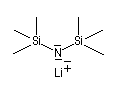
Les amines primaires et secondaires sont des acides de Brönsted très faibles pKa > 30. Contrairement aux alcools qu'on peut déprotoner, en quantité certes très faible, dans l'eau, la réaction n'est pas envisageable avec les amines. Les amines peuvent être déprotonées en milieu non aqueux par des bases très fortes telles que le butyllithium nBuLi. On prépare ainsi plusieurs bases lithiées. Par exemple le diisopropylamidure de lithium (iPr)2N-Li+, noté traditionnellement LDA.
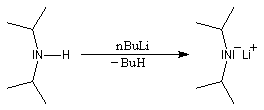
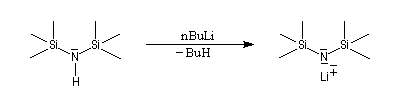
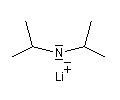 |
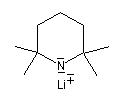 |
 |
|
LDA pK = 36 (solvant DMSO)
|
LTMP pK = 37 (DMSO)
|
LHMDS pK = 30 (DMSO)
|
- solubilité en milieu organique, même à basse température dans des solvants comme le THF ;
- grande force (pKa = 36) ;
- nucléophilie très faible du fait de l'encombrement important des groupes isopropyles.
Une autre méthode pour déprotoner les amines consiste à coupler la réaction acide-base avec la réduction de l'ion H+ grâce à un métal alcalin.
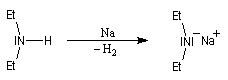
Complexation
Formation de complexes
Les amines sont des bases de Lewis et, à ce titre, forment de nombreux complexes avec les ions métalliques des éléments de transition. Les amines simples sont des ligands comparables à l'ammoniac. Une diamine comme l'éthylène diamine (diamino-1,2-éthane, symbolisée par én) possède deux points d'ancrage. C'est un ligand bidente (en anglais : bidentate.) L'augmentation très importante de la constante de stabilité observée quand on remplace l'ammoniac par l'éthylènediamine trouve son origine dans l'effet entropique. Dans la réaction de formation du premier complexe, le nombre de particules n'est pas modifié.
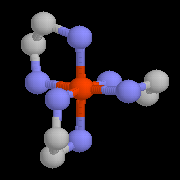 |
L'image de gauche représente un complexe du cobalt (III) utilisant l'éthylènediamine comme ligand [Co(en)3]2+. Ce complexe, préparé pour la première fois par A. Werner en 1912 présente la particularité d'être chiral. On trouve un complexe azoté du cobalt dans la partie centrale de la molécule de vitamine B 12 dont la synthèse totale a été effectuée par R. B. Woodward et A. Eschenmoser [48] |
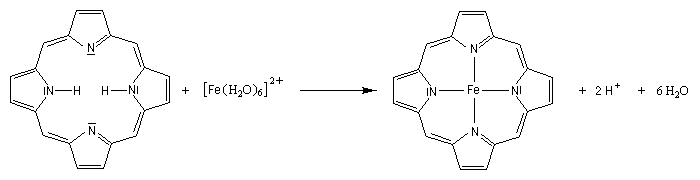
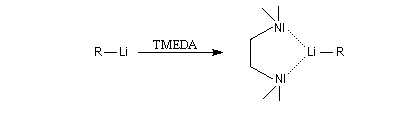
Les amines primaires et secondaires sont déprotonées quantitativement par les organomagnésiens et les organolithiens. Un exemple évoqué plus haut est la préparation de la base lithiée LDA. Les amines tertiaires sont compatibles avec les organométalliques et sont parfois utilisées comme solvant de ces composés. La tétraméthyléthylènediamine (TMEDA) forme des complexes stables, solubles en milieu organique avec les organolithiens. La complexation du lithium accroit fortement la polarité de la liaison organométallique et exalte le caractère de carbanion du lithien.
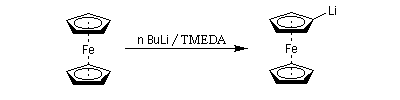
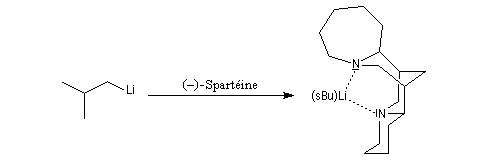
Les cryptands sont des macrohétérobicycles. On peut les considérer comme des structures prolongeant à trois dimensions celles des polyéthers macrocycliques de type couronne synthétisés par C. J. Pedersen en 1967. Ils sont capables de former des complexes stables et sélectifs avec un certain nombre de cations durs tels que les alcalins et alcalino-terreux. Ces complexes sont appelés cryptates.
![Cryptand [2,2,2]](http://www.faidherbe.org/site/cours/dupuis/images11/cryptand.gif) |
L'image de gauche représente le
cryptand [2,2,2] synthétisé par J.-M. Lehn et son équipe en 1969. Il
forme des complexes 1-1 avec
des cations métalliques que J.-M. Lehn a appelé cryptates du grec cryptos caché. La stabilité du complexe dépend de la nature du cation. Elle varie dans l'ordre : K+ > Rb+ > Na+ > Li+ > Cs+. Li+ et Cs+ sont respectivement trop petit et trop gros pour être complexés de façon efficace. J.-M. Lehn a obtenu le prix Nobel de chimie conjointement avec les Américains D. J. Cram et C. J. Pedersen en 1987. |
- La solubilité dans l'eau du sulfate de baryum est multipliée par 10 000 en présence du cryptand [2,2,2].
- Les cryptands sont aussi des agents de transfert de phase. La densité de charge d'un cation camouflé au sein d'un cryptate est fortement diminuée tandis que le caractère lipophile de l'ensemble est considérablement accru. L'anion associé peut être véhiculé en phase organique est faiblement lié au cryptate. De ce fait, sa réactivité s'en trouve exaltée. On peut ainsi réaliser des substitutions en utilisant l'ion fluorure comme nucléophile.
- L'aptitude de certains cryptands à complexer les cations alcalino-terreux a été mise à profit pour extraire le strontium (II) radioactif d'organismes vivants.
- Signalons pour terminer une réaction remarquable réalisée par
J. L. Dye et son équipe de l'Université du Michigan en 1974. En
dissolvant du sodium en présence de cryptand [2,2,2] ces auteurs ont
préparé un composé ionique de couleur jaune d'or contenant l'anion Na- !
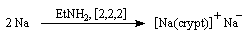
Alkylation
Bilan de l'alkylation d'Hofmann
La méthode a priori la plus simple pour alkyler l'atome d'azote d'une amine consiste à la faire réagir avec un dérivé halogéné. Avec une amine primaire ou secondaire il y a substitution d'un atome H par un groupe alkyle. On obtient donc en principe l'amine appartenant à la classe immédiatement supérieure. Avec une amine primaire, le bilan théorique s'écrit :
On peut s'affranchir de cette difficulté en utilisant une base peu nucléophile comme l'ion carbonate qui réagit avec les ions H+ formés.
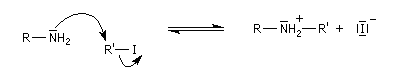
Mécanisme
Il s'agit d'une réaction de substitution utilisant comme substrat le dérivé halogéné et l'amine comme réactif nucléophile. Avec un substrat primaire ou secondaire la réaction est de type SN2. Les substrats tertiaires sont peu exploitables en raison de la réaction concurrente d'élimination. L'amine peut en effet jouer le rôle de base.
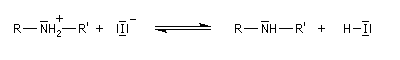
L'alkylation directe des amines avec un dérivé halogéné ne constitue généralement pas une très bonne méthode synthétique car l'amine alkylée réagit à son tour avec le réactif. On obtient donc un mélange de produits qu'il faut ensuite séparer. Une méthode simple et douce d'alkylation des amines est l'amination réductive des aldéhydes et des cétones.
La perméthylation d'Hofmann ou méthylation exhaustive, consiste à méthyler tous les sites possibles de l'amine. On utilise l'iodométhane comme agent alkylant. Avec ce substrat, la réaction est facilitée par le caractère d'excellent nucléoguge de l'ion iodure et il ne peut y avoir d'élimination. De plus, l'iodométhane est liquide à la température ordinaire. En présence d'un excès d'iodométhane, on obtient l'ion ammonium quaternaire.
|
n (mol)
|
3
|
2
|
1
|
|
Classe d'amine
|
primaire
|
secondaire
|
tertiaire
|
Remarque : la diméthylaniline peut être préparée en utilisant deux moles d'iodométhane pour une mole d'aniline.
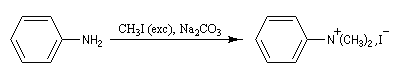
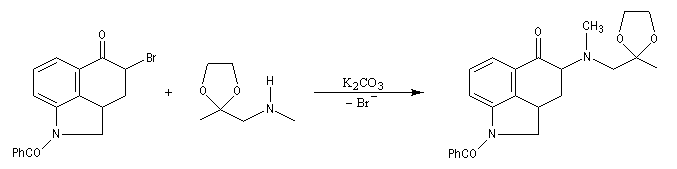
La réaction ci-dessous est extraite de la synthèse de l'acide lysergique (Woodward, 1956 [45]). Elle permet l'introduction d'une chaîne latérale en a du groupe carbonyle après bromation régiosélective dans cette position.
L'alkylation de l'ammoniac constitue une méthode de synthèse des amines primaires. Puisque l'ammoniac est très soluble dans l'eau, on peut utiliser des solutions aqueuses très concentrées. Le réactif est alors en grand excès par rapport à l'halogénure et la polyalkylation devient négligeable. La synthèse de la 1-méthyl-2-phényléthylamine illustre cette manière de procéder.
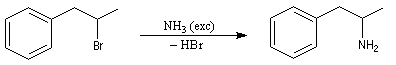
 |
Le terme amphétamine est le nom donné au mélange racémique de deux énantiomères de la 1-méthyl-2-phényléthylamine. Ils ont une structure voisine d'un neurotransmetteur : l'adrénaline. Ce type de composé agit comme stimulant du système nerveux central. Les composés de la famille des amphétamines ont été utilisé par certains sportifs dans le but d'améliorer leurs performances. Il sont sans doute à l'origine de la mort dramatique du champion du monde cycliste Tom Simpson dans l'ascension du mont Ventoux lors du Tour de France 1967. |
L'atome d'azote d'une amine primaire ou secondaire peut substituer un halogène porté par un cycle aromatique fortement désactivé selon un mécanisme d'addition-élimination.
Le 1-chloro-2,4-dinitrobenzène forme des dérivés facilement cristallisables permettant d'identifier les amines [6].
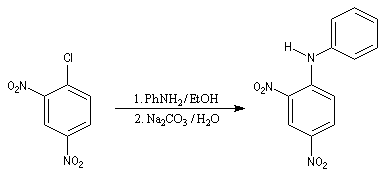
Réactions d'élimination à partir des ions ammonium quaternaires
Les ions ammonium quaternaire possèdent un groupe -NR3+ lié à une chaine carbonée. Si l'atome de carbone en b possède un atome d'hydrogène, une élimination conduisant à un composé éthylénique est possible.
Les amines étant d'assez mauvais nucléofuges (à corréler empiriquement à une basicité élevée) la réaction doit être effectuée en présence d'une base forte à chaud.
L'oxyde d'argent en suspension dans l'eau ou oxyde d'argent "humide", est très utilisé. Son rôle est double :
- C'est un oxyde basique, qui en présence d'eau, est une source d'ions OH- :
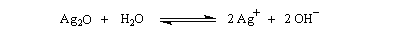
- L'ion Ag+ réagit avec avec I- pour donner de l'iodure d'argent AgI très peu soluble, ce qui décolle cet ion de la paire d'ions formée avec l'ion alkylammonium.
|
Composé
|
but-2-ène (Z et E)
|
but-1-ène
|
|
%
|
5
|
95
|
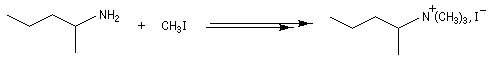

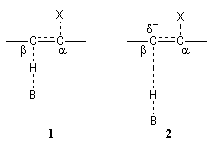 |
Le schéma de gauche représente une illustration de la théorie de l'état de transition variable. Dans le complexe activé (1) d'une élimination bimoléculaire E2 l'arrachage de l'atome d'hydrogène par la base B et le départ du nucléofuge X sont synchronisés. En présence d'un mauvais nucléofuge X, l'arrachage de l'atome d'hydrogène précède le départ de X. Il en résulte pour le complexe activé (2) l'apparition d'une charge négative sur l'atome de carbone en b qui lui confère un caractère de carbanion. |
A l'époque où l'on ne disposait pas du puissant moyen d'analyse que constituent les méthodes spectrales, la détermination de la structure des molécules s'effectuait par voie chimique. L'élimination d'Hofmann a été mise à profit pour déterminer la structure de molécules complexes notamment des alcaloïdes. La nature des composés éthyléniques issus de l'élimination d'Hofmann peut être déduite de l'analyse des produits de coupure par ozonolyse. Examinons un exemple. Soit à déterminer la position du groupe méthyle dans l'amine cyclique ci-dessous qui est un dérivé de la pipéridine.
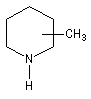
- ozonolyse ;
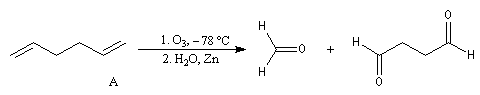
- méthylation ;
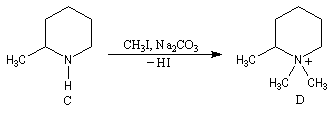
- élimination d'Hofmann ;
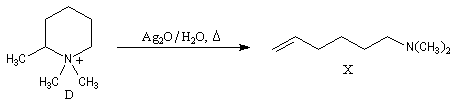
- méthylation ;
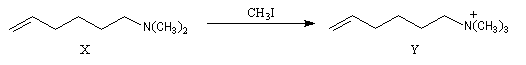
- élimination d'Hofmann ;
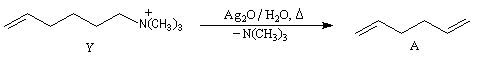
La réaction d'Hofmann est utilisée dans la synthèse de doubles liaisons éthyléniques.
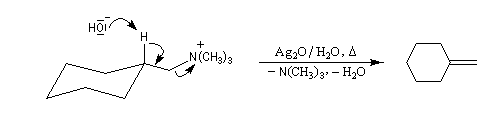
L'exemple suivant concerne les deux dernières étapes de la synthèse du barrelène par H. E. Zimmerman (1960).
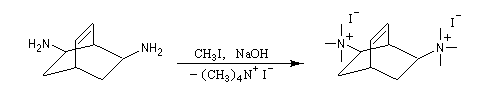
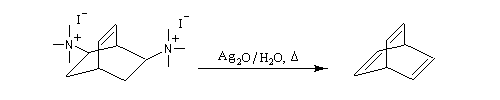
Utilisation des ions ammonium quaternaires dans les réactions par transfert de phase
Les ions ammonium quaternaires sont des amphiphiles. Ils comportent une partie apolaire et une partie ionique. Cette structure originale leur permet d'être solubles en milieu aqueux sous forme ionique et d'exister en milieu organique engagés dans des paires d'ions faiblement liés. Lorsqu'une réaction implique des espèces anioniques, l'une des difficultés est de disposer d'anions suffisamment réactifs dans la phase organique. Une solution à ce problème consiste à véhiculer les anions dans cette phase grâce à un contre-ion positif qui partage son affinité entre la phase aqueuse et la phase organique. Ces ions transporteurs sont recyclés au fur et à mesure de la réaction, c'est pourquoi on parle de catalyse par transfert de phase. Le schéma ci-dessous résume les principaux équilibres dans le cas d'une réaction de substitution :
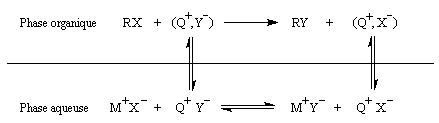
 |
La catalyse par transfert de phase a été découverte
par le chimiste polonais M. Makosza en 1965. Elle consiste à réaliser
une réaction dans un milieu diphasé organique / aqueux en présence d'un
catalyseur
pouvant se partager entre les deux milieux, comme un ion ammonium
quaternaire R4N+ un éther couronne ou un cryptand.
Cet ion peut véhiculer les anions en phase organique où ils acquièrent
une plus grande réactivité. La réaction peut être réalisée à température
assez basse sans utiliser de solvant dipolaire aprotique. Un site [14] est spécialement consacré à ce type de catalyse. |
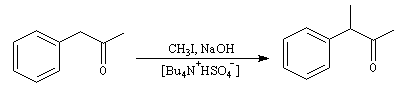
On peut aussi synthétiser des éthers en milieu biphasé organique/aqueux en utilisant les ions OH- comme base en présence d'un ammonium quaternaire comme agent de transfert de phase.
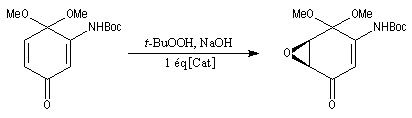
Bilan
Il s'agit de la réaction entre une amine primaire ou secondaire et un agent acylant : halogénure d'acyle, anhydride d'acide, acide carboxylique. Elle fournit un amide. On observe une réaction du même type avec l'ammoniac. La présence d'au moins un atome d'hydrogène sur l'azote est essentielle. C'est la raison pour laquelle les amines tertiaires ne peuvent être acylées.
Acides carboxyliques
La réaction entre une amine et un acide carboxylique engage l'essentiel des réactifs sous forme de sel d'aminium inerte vis à vis de la réaction d'acylation puisque cet ion a perdu tout caractère nucléophile.
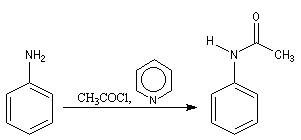
Chlorures d'acyles et anhydrides
La réaction entre une amine primaire ou secondaire et un chlorure d'acyle ou un anhydride permet la préparation des amides. La méthode la plus ancienne est celle de Schotten-Baumann. La réaction est effectuée en milieu aqueux avec une solution diluée d'ions hydroxyde OH- dont le rôle est de neutraliser l'acide formé. L'amine est suffisamment nucléophile pour que la réaction concurrente des ions hydroxyde vis à vis de l'halogénure d'acyle soit négligeable.
Au début du siècle, A. Einhorn a modifié le protocole initial en remplaçant les ions hydroxyde par la pyridine.
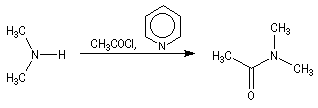
Plus récemment W. Steglich et L. M. Litvinenko ont introduit la diméthylaminopyridine (DMAP) qui s'est révélé un catalyseur extrêmement efficace dans ce genre de réactions.
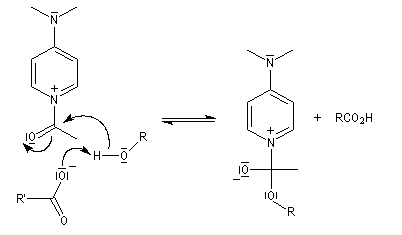
Mécanisme dans le cas des halogénures d'acyles
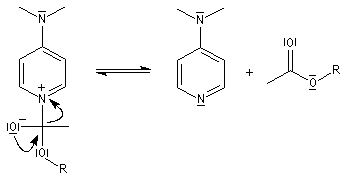
Il s'agit d'un mécanisme par addition-fragmentation. La réaction ressemble à celle vue avec les alcools. L'étape cinétiquement déterminante est la formation de l'intermédiaire tétraédrique. Ce dernier subit une fragmentation quasiment non renversable avec départ de l'ion halogénure.
- addition nucléophile ;
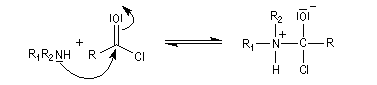
- fragmentation de l'intermédiaire tétraédrique ;
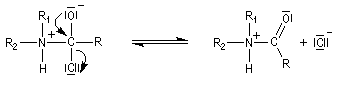
- l’ion acylaminium est ensuite déprotoné par une base comme la pyridine.
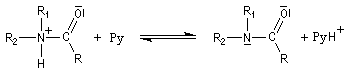
- elle neutralise l'acide formé ;
- il s'agit d'un catalyseur nucléophile qui forme un adduit intermédiaire (I) avec le chlorure d'acyle.
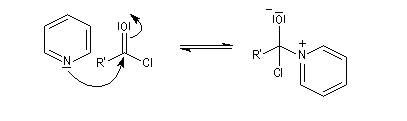
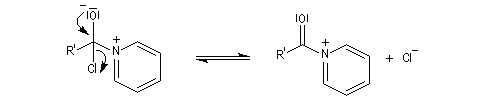

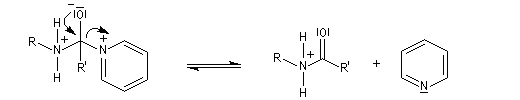
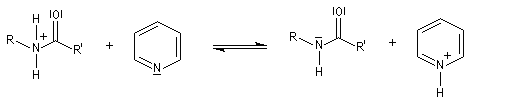
Catalyse par la DMAP La N, N-diméthylaminopyridine (DMAP), est utilisée pour accélérer les réactions d'acylation des alcools et des phénols ainsi que dans certaines hydrolyses. Il s'agit d'un catalyseur très efficace même dans les réactions intramoléculaires difficiles comme les macrolactonisations et les macrolactamisations. La DMAP est également utilisée comme catalyseur de la réaction de Baylis-Hillman.
 |
A la température ordinaire la DMAP se présente sous la forme d'un solide blanc, fondant à 111,6 °C. Il s'agit d'un composé toxique, à manipuler avec précautions. La DMAP est largement utilisée au laboratoire et en synthèse comme catalyseur nucléophile. |
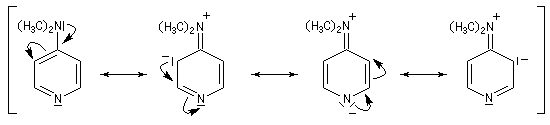
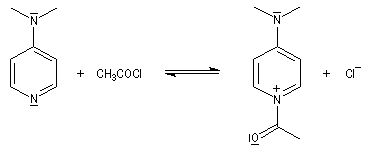
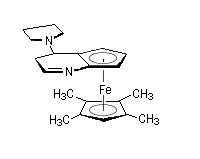 |
Des dérivés chiraux de la DMAP utilisant un groupement ferrocénique ont été préparés très récemment [31]. Ils servent de catalyseurs dans différents types de transformations :
|
Exemple : la synthèse de l'acide amino-3-benzoïque peut être effectuée par oxydation du groupe méthyle de la 3-méthylaniline mais les amines aromatiques étant très sensibles à l'oxydation, la réaction directe est impraticable. Il faut au préalable effectuer la protection du groupe amino :
- protection de la fonction amine ;
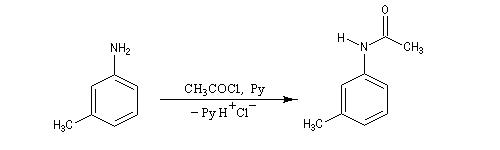
- oxydation du groupe méthyle ;
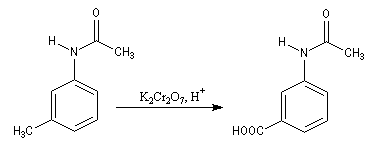
- régénération du groupe amino par hydrolyse de l’amide en milieu acide ou basique.
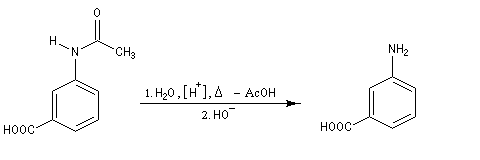
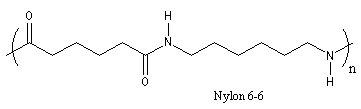
Le Nylon 6-6 est un polyamide artificiel. Sa synthèse a été réalisée par le chimiste américain W. H. Carothers (du Pont de Nemours). Dans l'industrie, on le prépare par réaction entre l'acide hexanedioïque (acide adipique) et le diamino-1,6-hexane (hexaméthylène diamine) à 280 °C.
 |
Le nylon est utilisé comme fibre textile ainsi que dans la fabrication de pièces mécaniques (paliers, engrenages etc.) Parmi les nombreuses anecdotes concernant l'origine du nom choisi par Carothers, l'une d'elles probablement apocryphe, est à mettre en relation avec le fait qu'à l'époque une bonne partie de la soie provenait du Japon. Nylon serait constitué des premières lettres de la phrase : Now You're Lost Old Nippons. |
L'acylation de la b-phényléthylamine par un chlorure d'acyle est la première étape d'une réaction permettant de préparer les quinolidines substituées connue sous le nom de réaction de Bischler-Napieralski. Il s'agit d'une synthèse mettant en jeu plusieurs étapes qui possède plusieurs points communs avec la réaction de Vilsmeier-Haack.
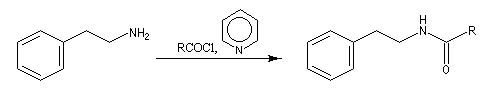
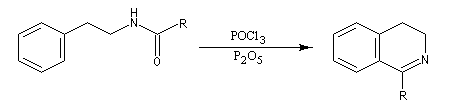
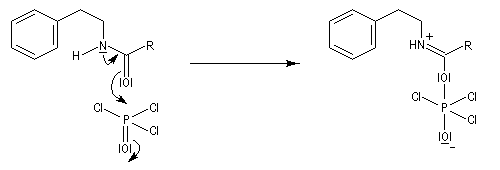
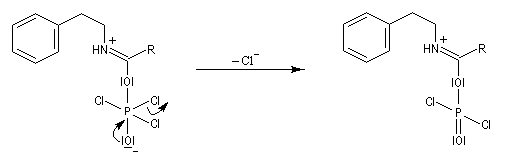
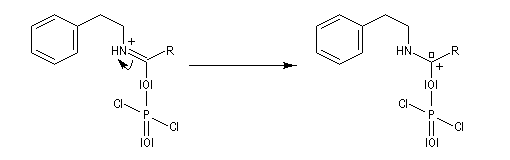
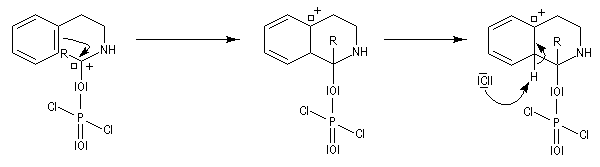
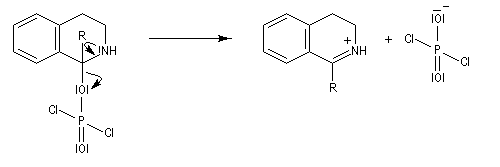
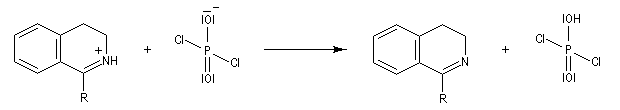
 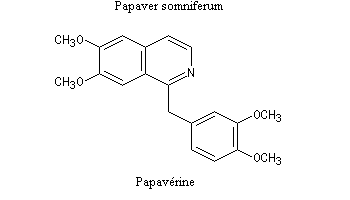 |
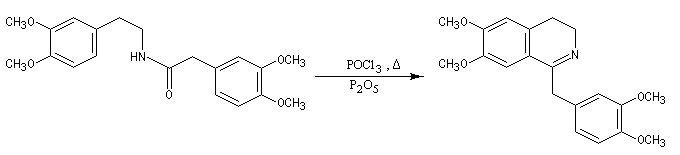
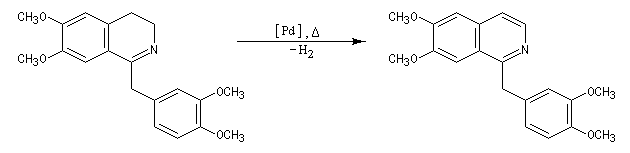
Lorsqu'on incise les capsules de pavot somnifère (papaver somniferum), celles-ci exsudent un liquide laiteux. Après séchage, il conduit à une résine brune qui constitue l'opium. L'opium contient de nombreux alcaloïdes : codéine, papavérine, laudanosine, morphine. La morphine (du grec Morphée : Dieu du sommeil) est l'alcaloïde le plus important de l'opium qui en contient jusqu'à 23 %. Isolée par F. Sertüner, sa structure a été établie en 1925 par J. M. Gulland, R. Robinson et C. Schöpf. La première synthèse de la morphine (racémique) a été réalisée en 1953 par J. M. Gates. Il s'agissait d'une synthèse linéaire comportant 29 étapes pour un rendement total de 0.0014 % ! [20]. 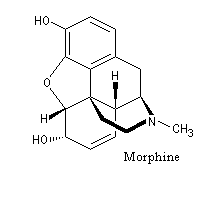 |
Bilan
La réaction entre un halogénure d'acide sulfonique et une amine primaire ou secondaire fournit une sulfonamide.
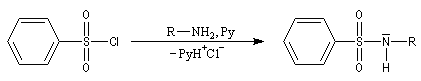
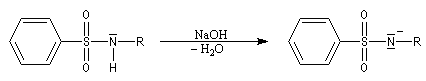
Test de Hinsberg
Ce test est basé sur la solubilité de la sulfonamide quand elle existe en milieu basique. Un mode opératoire pour la réalisation de ce test est détaillé à la référence [3]. Les résultats sont résumés dans le tableau ci-dessous.
|
Classe de l’amine
|
primaire
|
secondaire
|
tertiaire
|
|
Solubilité sulfonamide
|
oui
|
non
|
pas de sulfonamide
|
Imines
Synthèse des imines et des ions iminium
L'addition d'une amine primaire sur un composé carbonylé conduit, par une réaction équilibrée, à un amino-alcool peu stable. La réaction est catalysée en milieu acide. Cependant le pH ne doit pas être trop bas sinon l'amine est protonée et l'addition nucléophile ne se produit plus.
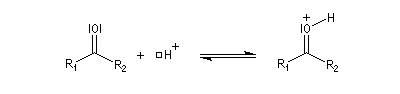
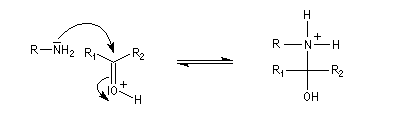
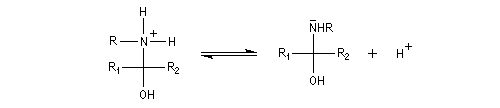
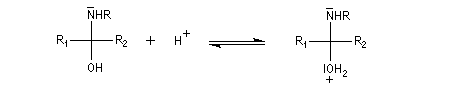
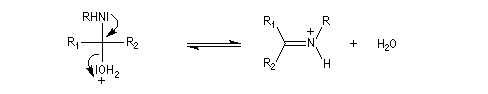

Les imines peuvent être regardées comme les analogues azotés des composés carbonylés. Elles possèdent comme ces derniers une liaison double polarisée. On peut donc prévoir qu'elles donneront lieu à des réactions d'addition. Les imines dérivant de l'ammoniac sont instables. Si l'on fait réagir le méthanal avec l'ammoniac on obtient un composé cristallin de couleur blanche : l'hexaméthylènetétramine (HMTA). Au cours de cette réaction, l'aldimine intermédiaire, instable, ne peut pas être isolée.
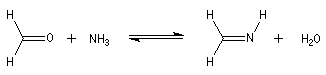
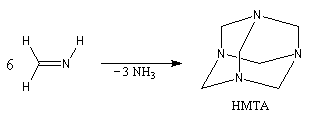
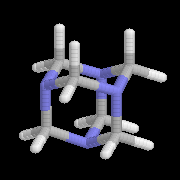 |
Le Tétraazatricyclo[1,1,3,3,7,7]décane ou hexaméthylènetétramine(HMTA) est un composé curieux rappelant la molécule d'adamentane. A la température ordinaire, il se présente sous la forme de cristaux blancs, solubles dans l'eau, qui fondent à 280 °C. L'HMTA est utilisé en médecine comme antiseptique sous le nom d'urotropine (étym : qui fait bouger l'urée) en raison de ses propriétés diurétiques. Il s'agit d'une source de cation iminium qui est utilisée dans la formylation de composés aromatiques activés (réaction de Duff) [46] |
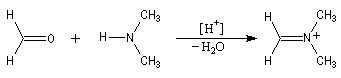
Les benzodiazépines sont des composés psychoactifs, anxiolytiques et hypnotiques. Elles sont utilisés notamment en anesthésie. L'analyse rétrosynthétique des benzodiazépines [1,4] montre qu'on peut les obtenir en créant une imine et un amide.

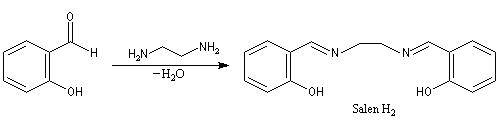
Ligand salen, complexes de Jacobsen
La synthèse du composé salen-H2 constitue un autre exemple de réaction de ce type. Cette imine joue le rôle de ligand vis à vis du cobalt (II) dans un complexe capable de fixer le dioxygène de façon réversible.
- l'époxydation énantiosélective des composés éthyléniques est réalisable en utilisant un complexe du Mn (III) et un ligand chiral de symétrie C2 dérivé du salen ;
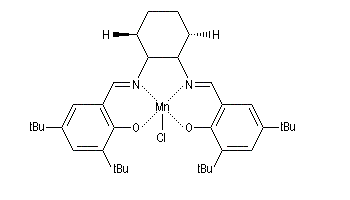
- l'ouverture énantiosélective d'époxydes avec [Cr(salen)].
La formation d'une imine suivie de son hydrogénation in situ, constitue une méthode d'alkylation douce des amines appelée amination réductive.
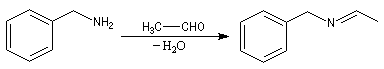
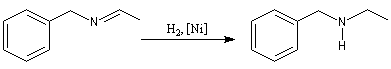

Addition des organométalliques
L'addition d'un organomagnésien sur une imine substituée suivie d'une hydrolyse de l'adduit constitue une méthode de synthèse d'amines secondaires.
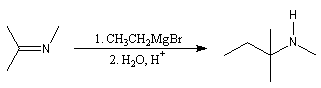
Il s'agit d'une méthylation des amines primaires et secondaires au moyen du mélange méthanal-acide méthanoïque (acide formique). La réaction procède en deux étapes. Raisonnons dans le cas d'une amine secondaire.
- Il se forme dans un premier temps un ion iminium.
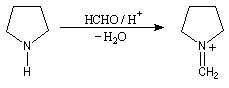
- L'acide méthanoïque réduit cet ion iminium par transfert d'un ion hydrure.
Finalement l'amine de départ a été méthylée sur l'atome d'azote.
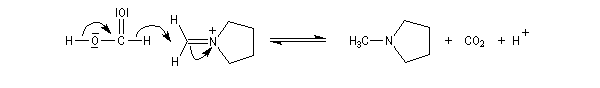
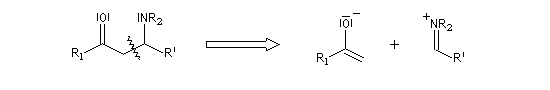
La réaction de Mannich consiste en l'addition d'un atome de carbone nucléophile sur un ion ion iminium formé in situ. Le réactif nucléophile est l'énol d'un composé carbonylé ou d'un phénol. La réaction est facilitée lorsque l'ion iminium est issu de la condensation entre le méthanal et une amine primaire ou secondaire car dans ce cas l'atome de carbone du réactif est très électrophile.
Le schéma rétrosynthétique est le suivant :
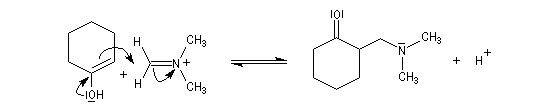
- formation de l'ion ion iminium (sel d' Eschenmoser) ;
- équilibre de tautomérie entre le composé carbonylé et son l'énol ;
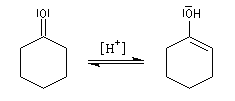
- réaction d'addition entre l'énol et l'ion ion iminium.
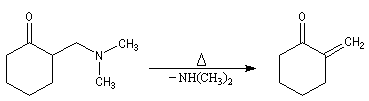
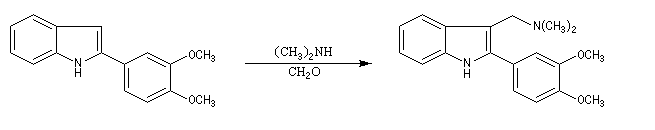
La réaction de Mannich a été utilisée dans de nombreuses synthèses de composés appartenant à la famille des alcaloïdes. L'exemple suivant est une étape de la synthèse de la strychnine par R. B. Woodward [38]
La tropinone est une molécule bicyclique dont on retrouve le squelette dans de nombreux alcaloïdes. On peut l'obtenir par dégradation de l'atropine présente à l'état naturel dans la belladone.
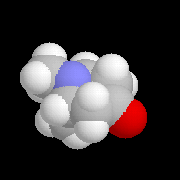 |
L'hyosciamine est l'un des principaux alcaloïdes d'une plante herbacée appelée atropa belladonna
(Belladone). Ses décoctions possèdent la propriété de dilater la
pupille de l'oeil. Elles étaient utilisées au XVIII ème siècle par les
femmes italiennes pour augmenter la profondeur de leur regard. Le nom de
Belladone vient de bella donna qui signifie jolie femme en italien. Le mélange racémique de l'hyosciamine et de son énantiomère est appelé atropine.
Il est utilisé comme antidote de certains gaz de combat car il agit
comme inhibiteur des récepteurs parasympathiques. Son mécanisme d'action
s'explique par sa fixation sur les récepteurs de l'acétylcholine dans
le système nerveux central et périphérique. C'est le médicament de
référence contre le malaise vagal.
|
Au cours de son étude remarquable des alcaloïdes qui devait le rendre mondialement célèbre et lui valoir le prix Nobel de chimie en 1947, le chimiste britannique R. Robinson (Université d'Oxford, [28]) en a donné une synthèse en 1917 qui est devenue un classique. Elle est basée sur une double réaction de Mannich. Il s'agit d'un exemple de réaction domino (ou réaction en cascade) car les évènements se déroulent les uns à la suite des autres sans qu'il soit nécessaire d'isoler un composé intermédiaire [43].
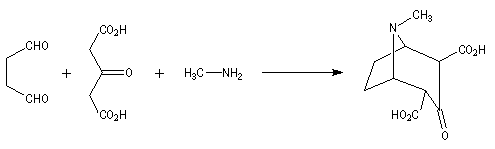
La réaction entre le succinaldéhyde et la méthylamine fournit un aminoalcool qui se cyclise spontanément.
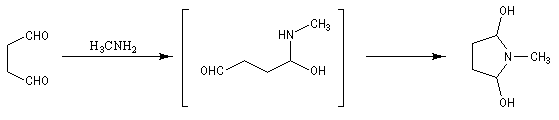
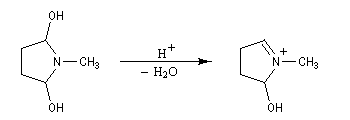
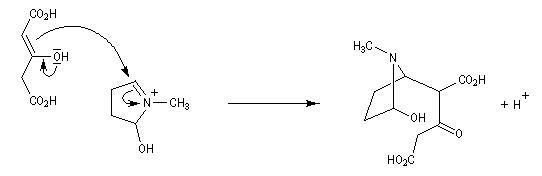
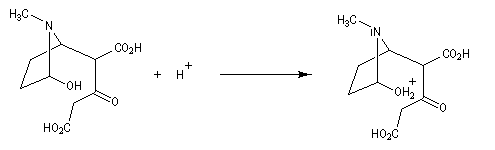
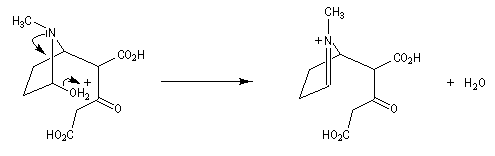
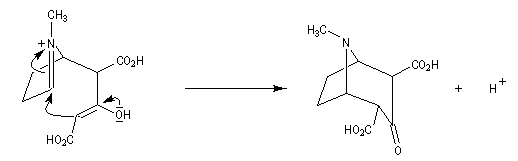
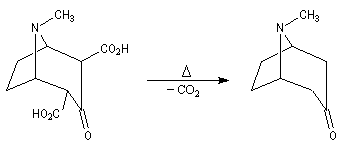
Origine
Les énamines peuvent être considérées comme les analogues azotés des énols et des éthers d'énols. Les énamines possédant un atome d'hydrogène sur l'azote (énamines primaires et secondaires) sont peu stables et se réarrangent en imines.
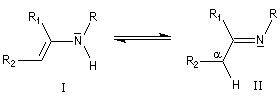
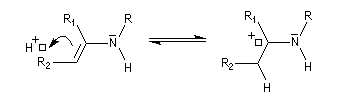
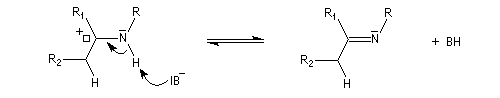
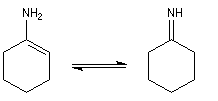
On prépare les énamines par réaction entre une amine secondaire et un composé carbonylé possédant un atome d'hydrogène sur l'atome de carbone en a du carbonyle en présence d'un catalyseur comme l'APTS. Le mécanisme suivant concerne la synthèse d'une énamine tertiaire en présence d'un catalyseur noté BH+.
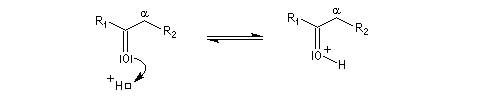
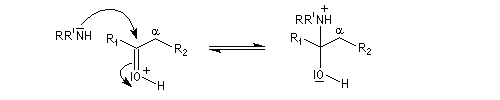
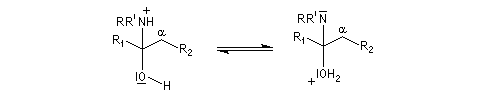
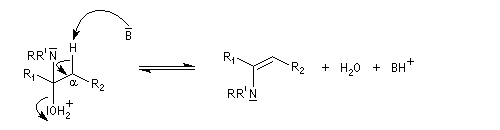
Les énamines sont stables en milieu basique et peuvent être utilisées dans certains cas comme groupement protecteur de la fonction carbonyle. Le traitement de l'énamine par un excès d'eau en présence d'une quantité catalytique d'acide fournit l'amine et le composé carbonylé parents. L'exemple ci-dessous concerne la réaction entre la cyclohexanone et la pyrrolidine.
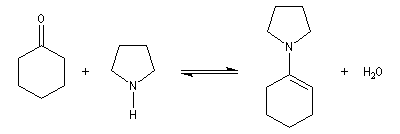
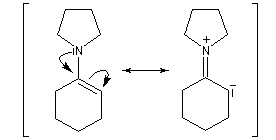
Acylation des énamines
La réaction entre une énamine et un agent acylant assez réactif comme un chlorure d'acyle permet la synthèse de systèmes dicarbonylés 1, 3. Un mode opératoire de la réaction suivante se trouve à la référence [12].
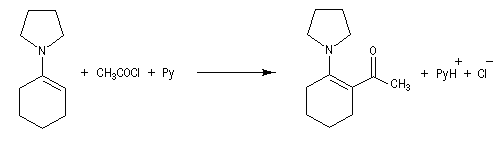
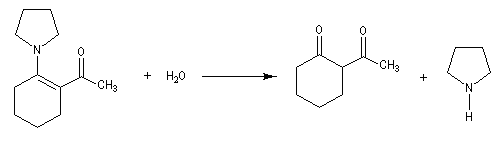
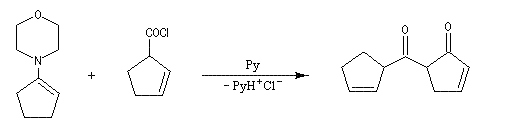
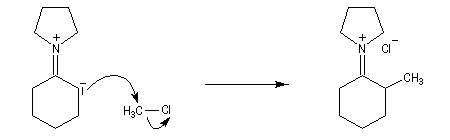
La méthylation des cétones sur l'atome de carbone en a du carbonyle pose un problème de régiosélectivité. La réaction par union directe de la cétone et d'un réactif alkylant comme l'iodométhane fournit un mélange de cétone mono et polysubstituée. Pour pallier cette difficulté plusieurs méthodes ont été proposées. L'une d'elles, mise au point par le chimiste américain d'origine belge G. Stork, consiste à utiliser une énamine comme intermédiaire.
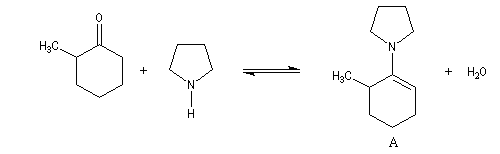
Une autre méthode d'alkylation régiosélective des cétones consiste à mettre à profit les propriétés des éthers de silyle. Régiosélectivité
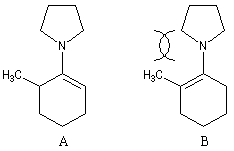
La réaction entre la méthylcyclohexanone et la pyrrolidine fournit l'énamine possédant la liaison double la moins substituée (A) à l'exclusion de son isomère (B). La régiosélectivité de l'élimination est donc opposée à celle qu'on aurait si la règle de Zaytsev était suivie.
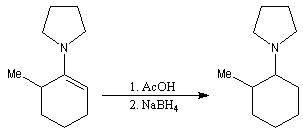
Les énamines peuvent être réduites par hydrogénation catalytique. NaBH4 ne réagit pas directement avec elles. En revanche ce réactif réduit rapidement les ions iminium obtenus en traitant l'énamine en milieu acide.
Formation de l'électrophile
La nitrosation est la réaction entre une amine et l'acide nitreux HNO2. Ce dernier est un composé instable vis à vis de sa dismutation en NO3- et NO. Il est préparé dans le milieu réactionnel en acidifiant une solution de nitrite de sodium par l'acide chlorhydrique. La véritable entité électrophile est le cation nitrosonium NO+ qui est impliqué dans les équilibres suivants :
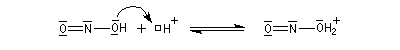
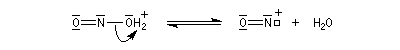
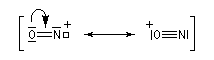
Amines secondaires
On obtient une N-nitrosoamine.
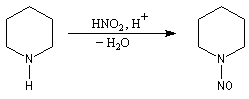
Amines primaires Il se forme un ion diazonium.
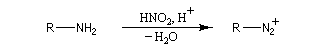
- Les
ions diazonium aliphatiques sont très peu stables car le diazote est un excellent nucléofuge. On obtient un carbocation susceptible de donner lieu à plusieurs réactions selon la nature des réactifs présents dans le milieu réactionnel.
Notons que ces sels de diazonium sont explosifs à l'état solide.
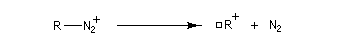
- Les ions diazonium aromatiques donnent lieu à de nombreuses réactions qui sont étudiées dans un chapitre particulier.
Activation, régiosélectivité
L'aniline réagit beaucoup plus vite que le benzène dans les réactions de substitutions électrophiles. Le groupe -NH2 est activant et oriente en ortho-para.
Bromation
La bromation de l'aniline peut s'effectuer très facilement. Le groupe amino est tellement activant qu'il suffit de mélanger l'aniline et une solution aqueuse de brome, pour obtenir immédiatement un précipité blanc de tribromoaniline.
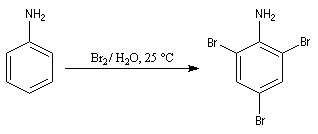
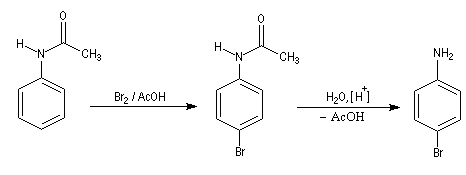
La nitration directe de l'aniline est impossible car l'acide nitrique oxyde le groupe amino. Pour s'affranchir de cette difficulté, on utilise un groupe protecteur.
- On commence par préparer la
N-phényléthanamide par réaction entre l'aniline et le chlorure
d'éthanoyle, ce qui permet la protection du groupe amino.
On effectue ensuite la nitration de la N-phényléthanamide. Le groupe
acétamido oriente la substitution électrophile en ortho et para. Des deux isomères qui peuvent a priori se former, le dérivé para est majoritaire
car la position ortho est plus encombrée que la position para.
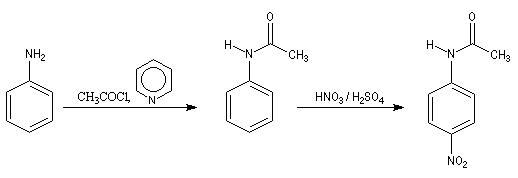
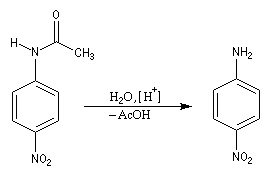
- Il ne reste plus qu'à hydrolyser l'amide en milieu acide ou
basique afin de régénérer le groupe amino. La 4-nitroaniline est un
solide de couleur jaune (pF = 148 °C).
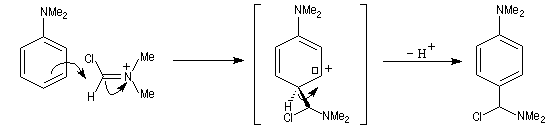
Avec les amines tertiaires aromatiques, l'acide nitreux en milieu acide conduit à une substitution électrophile sur le cycle. L'entité électrophile est l'ion nitrosonium :
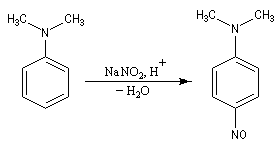
le mécanisme est le suivant :
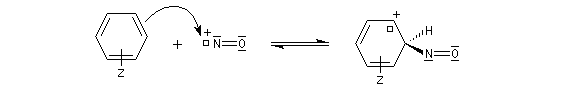
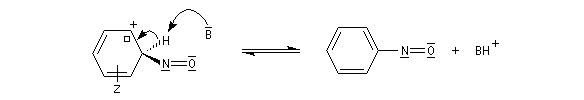
Rappelons qu'avec les amines primaires, l'action de l'acide nitreux conduit à union diazonium. Avec les amines secondaires, on obtient des N-nitrosoamines. Un mode opératoire est décrit à la référence [6]. Formylation de Vilsmeier-Haack La réaction de Vilsmeier-Haack constitue une méthode de formylation des amines aromatiques N, N-disubstituées et de certains hétérocycles comme le pyrrole. On peut la regarder comme une extension de la réaction de Mannich. La préparation du réactif de Vilsmeier est donnée ci-dessous.
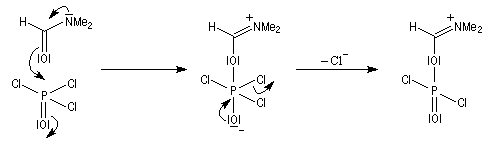
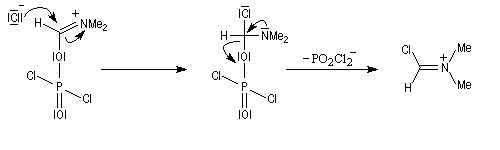
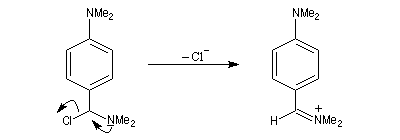
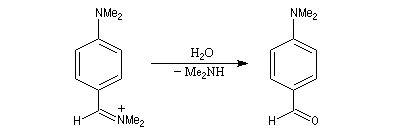
Oxydation des amines tertiaires
Les amines sont des composés assez sensibles à l'oxydation. Les produits de réaction dépendent de la classe de l'amine et de la nature de l'oxydant utilisé. La réaction la plus importante concerne les amines tertiaires. Les oxydants utilisés sont le mCPBA, peroxyde d'hydrogène ou encore l'acide peroxymonosulfurique (acide de Caro).
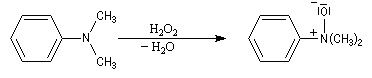
Les réactions importantes des oxydes d'amines sont étudiées dans un chapitre particulier :
Oxydation de l'aniline
L'oxydation de l'aniline dépend de l'agent oxydant utilisé :
- Avec CrO3, on obtient la benzoquinone.
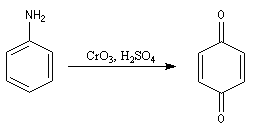
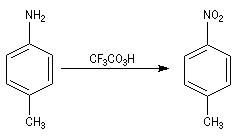
- On peut aussi utiliser un peroxyacide comme CF3CO3H, le groupe amino est oxydé en groupe nitro.
Bibliographie Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education vol 55, 1977.
[12] S. Hünig, E. Lücke, W. Brenninger, Organic Syntheses, V, p 533.
Liens
[13] The molecule of the month
[14] Phase Transfer Catalysis home page.
[15] Tétrodotoxin by J. Johnson - Florida State University
[16] La belladone
[17] Hemlock poison
[18] Introduction à la chimie supramoléculaire Teodor Silviu Balaban, Université d' Aix-Marseille III, „Paul Cezanne“; Karlsruhe Institute of Technology
[19] Mauveine by H. Rzepa
[20] Synthesis of Morphin Alkaloids
[21] R. B. Woodward - The Nobel Prize in chemistry 1965
[22] Biologie moléculaire Cours de D. Gautheret, Université de Paris Sud
[23] Preparation of trancyclooctene by Arthur C. Cope and Robert D. Bach, Massachusetts Institute of Technology, Cambridge 39, Massachusetts
[24] Preparation of methylenecyclohexane by Arthur C. Cope and Engelbert Ciganek, Massachusetts Institute of Technology, Cambridge 39, Massachusetts
[25] R. B. Woodward speaking on cephalosphorin C
[26] R. B. Woodward by James B. Hendrickson, who is Professor of Chemistry at Brandeis University, Waltham, Massachusetts
[27] R. Willstätter, The Nobel Prize in Chemistry 1915
[28] R. Robinson, The Nobel Prize in Chemistry 1947
[29] 1-Morpholino-1-cyclohexene by Hünig, E. Lücke, and W. Brenninger
[30] The Nobel prize in physiology and Medicine 1962
[31] The Fu research Group
[32] Optical isomers of Tris(ethylenediamine)cobalt (III)
[33] Synthèses de médicaments anticancéreux
[34] Rearrangement du squelette de la catharanthine
[35] Synthetic approaches to quinine
[36] Quinine, Definately
[37] Pierre Potier, médaille d'or du CNRS
[38] Synthèse totale de la strychnine
[39] Molecular Structure of Deoxypentose Nucleic Acids by Wilkins, M. H. F., Stokes, A. R., & Wilson, H. R. Nature 171, 738-740 (1953)
[40] Nomenclature des amines IUPAC, Gold Book.
Ouvrages théoriques
A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
J. March - Advanced organic chemistry, Wiley Interscience
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
P. Atkins - General Chemistry, Scientific American Books W. H. Freeman and Co.
J. Koolman, K. H Röhm - Atlas de biochimie, Médecine & Sciences, Flammarion.
P. Caubère - La catalyse par transfert de phase et son utilisation en chimie organique, Masson 1982.
Organic Synthesis Highlights V, Edited by Hans-Günther Schmalz and Thomas Wirth, J. Wiley 2003.
Articles
[40] C. F. Lane, Synthesis, 1975, 135-146
[41] G. Stork, Tetrahedron, 1982, 38, 1975, 3363
[42] Produits naturels anticancéreux, Daniel Guénard, Françoise Guéritte, Pierre Potier, l'actualité chimique, avril-mai 2003.
[43] Robinson, R. J. Chem. Soc. 1917, 111, 762-76 .
[44] Quinine : extraction, caractérisation, utilisation, Laibe-Darbour, Florence ; Aronica, Christophe Bulletin de l'Union des Physiciens, mars 2009, N° 912 p. 311-320
[45] Edmund C. Kornfeld, E.J. Fornefeld, G. Bruce Kline, Marjorie J. Mann, Dwight E. Morrison, Reuben G. Jones and R.B. Woodward.
[46] Duff, J. C.; Bills, E. J. J. Chem. Soc. 1932, 1987
[47] A Structure for Deoxyribose Nucleic Acid J. D. Watson and F. H. C. Crick (1) April 25, 1953 (2), Nature (3), 171, 737-738.
[48] The total synthesis of vitamin B12 R. B. Woodward Pure Appl. Chem., 1973, Vol. 33, No. 1, pp. 145-178









0 commentaires:
Enregistrer un commentaire