Cours de chimie organique -
Introduction à la chimie des composés organométalliques
Introduction
Origine
Un composé organométallique peut être défini comme un composé dans lequel il existe une liaison métal-carbone. Historiquement, le premier composé répondant à cette définition est un complexe du platine obtenu par le pharmacien danois W. C. Zeise en 1825. On peut le préparer en faisant barboter de l'éthène dans une solution de tétrachloroplatinate (II) de potassium.
L'analyse au rayons X révèle que le composé éthylénique est perpendiculaire au plan formé par l'atome de platine et les atomes de chlore.
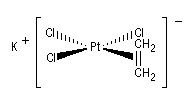
Classification des composés organométalliques
Un traitement correct de la liaison métal-carbone utilise la méthode des orbitales moléculaires. Nous utiliserons la classification simplifiée ci-dessous qui utilise un vocabulaire emprunté à la méthode du lien de valence :
- composés s, la liaison entre carbone et métal fait intervenir une ou plusieurs liaisons s. Dans
cette catégorie on trouve la plupart des organométalliques les plus
classiques, c'est à dire les organolithiens, les organomagnésiens, les
organocadmiens etc.
- composés p, la liaison entre carbone et métal fait intervenir une ou plusieurs liaisons p. A cette catégorie appartiennent les complexes p-éthyléniques, le ferrocène et les autres métallocènes.
Un autre exemple est constitué par les p-allyl-palladium (II) (h3-allyl PdX ; X = halogène, OAc,
OCOOR,…etc.)
Le tableau ci-dessous permet de classer les organométalliques selon le pourcentage de caractère ionique de la liaison C-métal.
Elément
|
K
|
Na
|
Li
|
Mg
|
Al
|
Zn
|
Cd
|
Pb
|
Hg
|
Cu
|
Electronégativité
|
0,82
|
0,93
|
0,98
|
1,31
|
1,61
|
1,65
|
1,69
|
1,87
|
2,00
|
2,5
|
% Caractère ionique
|
51
|
47
|
43
|
35
|
22
|
18
|
15
|
12
|
9
|
0
|
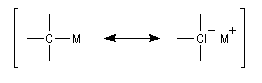
Les composés organosodiques forment de véritables carbanions.
Organomagnésiens Préparation
Le chimiste français V. Grignard synthétisa en 1901 le premier composé organomagnésien en faisant réagir l'iodure d'isobutyle et le magnésium dans l'éther ordinaire anhydre ou éthoxyéthane [1].
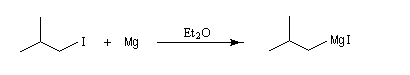
Les organomagnésiens iodés, bromés et chlorés sont obtenus la plupart du temps par insertion du magnésium dans la liaison carbone-halogène, en présence d'un solvant donneur d'électrons, le plus souvent un éther comme l'éthoxyéthane ou le THF anhydre.
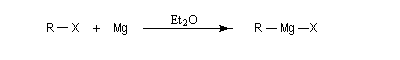
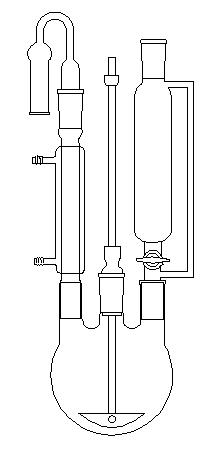 |
Le dessin ci-contre représente un montage classique pour la synthèse d'un composé organomagnésien. Le ballon tricol est surmonté d'un réfrigérant ascendant (à gauche) muni d'un tube desséchant à CaCl2 afin d'éviter l'entrée d'air humide dans le dispositif. Le dérivé halogéné est introduit par l'ampoule de coulée isobare située à droite. Un agitateur mécanique. (au centre) permet d'améliorer le contact entre les réactifs. On peut éliminer le risque de réaction secondaire entre l'organomagnésien et l'oxygène de l'air en effectuant un balayage du dispositif par un gaz inerte comme N2. Dans ce cas, le gaz inerte est introduit par le haut de l'ampoule de coulée. Le tube à CaCl2 n'est plus nécessaire. |
 |
La photographie représente la mise en œuvre du schéma précédent dans un montage pratique. L'ensemble du dispositif est surélevé par rapport à la paillasse de façon à pouvoir intervenir sous le ballon. Les différents éléments sont fixés au support vertical à l'aide de pinces. Le tube qui surmonte le réfrigérant contient du chlorure de calcium. Le support élévateur permet de placer un bain-marie pour augmenter la température du ballon en début de réaction. On peut, si c'est nécessaire, le remplacer rapidement par un cristallisoir d'eau froide pour évacuer plus rapidement la chaleur si la réaction devient trop rapide. Pour cette synthèse du magnésien du bromoéthane j'ai utilisé de l'éther séché sur de petits morceaux de sodium. Avec un dérivé bromé primaire comme le bromoéthane la réaction s'effectue très facilement si les différents réactifs sont bien secs. Au bout de quelques minutes l'ébullition de l'éther devient vigoureuse. On peut évacuer la chaleur avec un bain d'eau froide afin de diminuer la vitesse de réaction. L'éthoxyéthane est très volatil, très inflammable et sa vapeur est plus dense que l'air. Il est prudent de travailler avec de faibles quantités et de proscrire tout appareil générant des étincelles et bien sûr toute flamme. |
Lorsque la réaction a démarré, le dérivé halogéné introduit goutte à goutte à partir de l'ampoule de coulée. La réaction entre l'organométallique et le substrat qui a servi à le préparer est souvent une réaction indésirable appartenant à la même catégorie que la réaction de couplage de Wurtz.
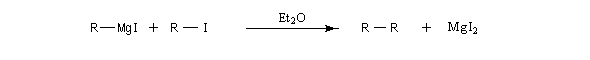
Le magnésium disparaît complètement en fin de réaction si les réactifs ont été introduits en proportions stœchiométriques. Avec du magnésium ultra pur, la solution est incolore. Le plus souvent on obtient un mélange gris foncé à cause des impuretés du métal.
La réactivité des halogénures d'alkyles décroît dans le même ordre que la mobilité de la liaison carbone-halogène.
RI > RBr > RCl >> RF
Les bromures d'alkyle ont une réactivité un peu plus faible que celle
des iodures. On préfère généralement utiliser les bromures en raison de
leur coût moins élevé sauf dans le cas de CH3I qui présente l'avantage d'être le seul dérivé méthylé liquide à la température ordinaire.L'ajout de quelques gouttes de dibromoéthane permet quelquefois de décaper le magnésium. Il s'agit d'une réaction d'élimination intramoléculaire.
R. D Rieke et S. E. Bales ont mis au point en 1974 une méthode de préparation de magnésium en poudre, très réactif par réduction de MgCl2 avec un métal alcalin en présence de KI [3].
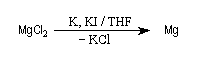
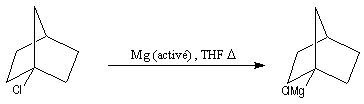

Les organomagnésiens alcyniques sont préparés par réaction entre un organomagnésien et un alcyne terminal.
Le caractère fortement réducteur du magnésium rend impraticable la préparation d'organomagnésiens fonctionnalisés par réaction entre un dérivé halogéné et le métal. Les magnésiens aromatiques portant des groupes fonctionnels comme le groupe ester peuvent être préparés avec succès grâce à une réaction d'échange métal-halogène dans des conditions très douces.
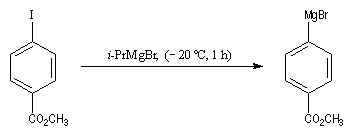
Les organomagnésiens de type allylique sont préparés assez facilement car la liaison métal-halogène des composés allyliques est très réactive.
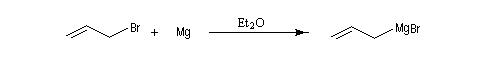
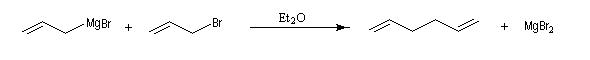
Les organomagnésiens et les organolithiens ont un atome métallique électrophile. Ce sont des acides de Lewis. La base de Lewis constituée par un composé donneur d'électrons va interagir avec l'organométallique et le stabiliser. Les éthers sont parmi les composés les plus couramment utilisés mais on peut aussi utiliser des amines tertiaires comme la triéthylamine. Mais lorsque la basicité de Lewis du composé croit, la réactivité de l'organométallique diminue.
Structure à l'état solide La plupart du temps, les magnésiens sont utilisés immédiatement après avoir été synthétisés. On peut toutefois les conserver à l'état sec, à l'abri de l'humidité et de l'air. Il faut faire attention que même à sec, il reste des molécules d'éther complexées avec l'organomagnésien et ce complexe peut s'enflammer spontanément dans l'air.
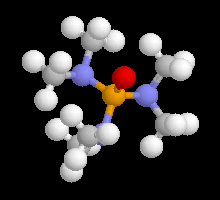
Avec les éthers, on a pu mettre dans certains cas en évidence à l'état solide, la présence de deux molécules d'éther par molécule d'organométallique.
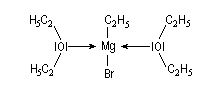
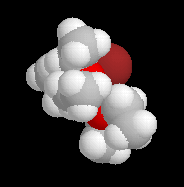 |
La structure d'un certain nombre de composés organomagnésiens à l'état solide a été déterminée par diffraction des rayons X. Dans [EtMgBr(OEt2)2] le magnésium est au centre d'un tétraèdre irrégulier. Sur l'image de gauche l'atome de magnésium est représenté en rose, celui de brome en marron. Les atomes d'oxygène sont en rouge. |
En solution, la situation est plus compliquée. Plusieurs équilibres interviennent simultanément et leur position dépend de plusieurs facteurs (concentration, température, solvant, etc.)
On peut mettre en évidence un équilibre entre magnésien mixte et symétrique, appelé équilibre de Schlenk. Dans le dioxane cet équilibre est déplacé en faveur du magnésien symétrique car dans ce solvant, MgX2 qui est peu soluble, précipite.
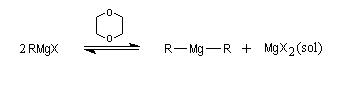
Le choix du solvant dépend notamment de la réactivité de l'halogénure. Avec les dérivés halogénés ordinaires, on utilise le plus souvent l'éther ordinaire ou éthoxyéthane. Dans le THF, les doublets de l'oxygène sont davantage dégagés stériquement que dans l'éthoxyéthane. Cet éther est ainsi plus solvatant que le précédent. Comme sa température d'ébullition est aussi plus élevée, il permet la synthèse d'organomagnésiens à partir de substrats peu réactifs comme les composés vinyliques. La préparation du THF anhydre est vue dans le chapitre relatif aux éthers.
Le caractère d'acide de Lewis des organomagnésiens se manifeste par la formation de structures chélatées avec certains composés oxygénés. C'est le cas par exemple avec les composés carbonylés substitués en a par un groupe alkoxy. Ces structures chélatées jouent un rôle important dans le déroulement stéréochimique des réactions des organomagnésiens sur ces composés.
Mécanisme de formation des organomagnésiens
Bien qu'il ait fait l'objet d'un nombre très important d'études, le mécanisme de formation des organomagnésiens n'est pas connu avec certitude. Globalement il y a eu insertion de magnésium dans la liaison carbone-halogène. Il s'agit donc d'une addition oxydante. Il est vraisemblable que le mécanisme implique une réaction en chaîne avec intervention de radicaux.
La liaison C- Mg possède un caractère ionique de 35 %. Cela confère aux réactifs de Grignard une position centrale dans la famille des organométalliques.
Basicité
Sont déprotonés de façon quantitative les fonctions suivantes : acides carboxyliques, phénols, alcools, amines, amides non substitués, alcynes terminaux. Lorsqu'un orgnomagnésien réagit sur un substrat porteur de l'une de ces fonctions, celle-ci doit être protégée. Il existe une grande variété de groupes protecteurs. A titre d'exemple, la fonction alcool peut être protégée par réaction avec un éther d'énol tel que le DHP.
La réaction entre un organomagnésien et un alcyne terminal est à la base de la préparation des magnésiens alcyniques. Certains atomes d'hydrogène acides portés par des atomes de carbone en a de fonctions carbonyles peuvent aussi être arrachés, notamment dans le cas des composés dicarbonylés qui possèdent des atomes d'hydrogène d'acidité notable. Les solvants très solvatants comme l'hexaméthylphosphotriamide (HMPT) favorisent l'énolisation des composés carbonylés.
Avec les magnésiens l'équation s'écrit :
Dosage des organomagnésiens & des organolithiens
La réaction entre un organomagnésien ou un organolithien et un alcool comme le butanol est quantitative. Cette réaction peut être donc mise à profit pour effectuer le dosage de l'organométallique.
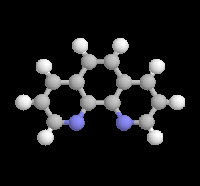 |
L'orthophénantroline-1,10 (image de gauche) et la bisquinoléine jouent, grâce à leurs atomes d'azote, le rôle de base de Lewis vis à vis du métal électrophile des magnésiens et des lithiens. Ces composés forment des complexes de transfert de charge colorés avec ces organométalliques : le bromure de phénylmagnésium dans l'éther donne un complexe de couleur rose saumon, le butyllithium dans le toluène donne un complexe de couleur rouge. |
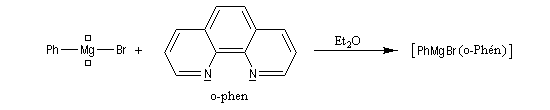
 |
La photographie de gauche représente une expérience destinée à mettre en évidence le rôle de l'orthophénantroline comme indicateur dans le dosage des organomagnésiens et des organolithiens. L'ajout d'une petite quantité de bromure de phénylmagnésium à une solution d'orthophénantroline dans l'éther fournit une coloration rose saumon. Cette coloration est due à la formation d'un complexe de transfert de charge entre l'orthophénantroline qui joue le rôle de donneur d'électrons et l'organomagnésien qui joue le rôle d'accepteur d'électrons. |
La méthode dite de Zérévitinov est assez ancienne. L'organomagnésien est mis à réagir avec une solution de concentration connue d'un composé à hydrogène mobile dissous dans un solvant inerte. On obtient un alcane dont on mesure le volume. Inversement, avec une solution titrée d'organomagnésien, on peut doser des composés à hydrogène mobile. Une réaction du même type, permet l'introduction de deutérium en utilisant de l'eau lourde.
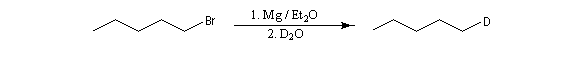
Cette réaction introduite par le chimiste japonais Kumada en 1972 constitue l'un des premiers exemples de réaction de couplage croisé [35]. Les partenaires sont un organomagnésien et un dérivé halogéné. Elle est catalysée par un complexe du nickel tel que NiCl2(dppe) ; (dppe : diphénylphosphinoéthane) [23].
R1 : alkyle, alcényle, aryle
X : halogène (I, Br) , OTf (triflate) ;
R2 : alkyle, alcényle, aryle
X : halogène (I, Br, Cl)
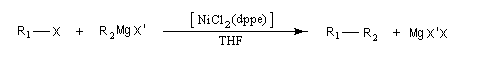
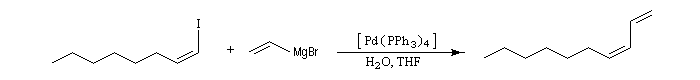
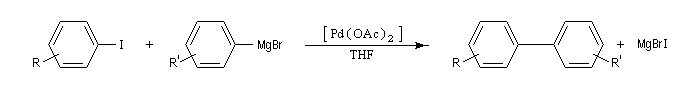
remarque : le couplage des organomagnésiens avec les dérivés halogénés peut être catalysé par Fe(DBM)3 (DBM = dibenzoyle méthane). Cette réaction est appelée couplage de Kochi [37].
Réactions avec des réactifs inorganiques
Carbonatation des organomagnésiens et des organolithiens
Ces réaction ont été étudiée dans le chapitre sur les dérivés d'acides.
Dioxygène, soufre
La réaction entre un organomagnésien et le dioxygène en excès à basse température (- 78 °C dans le mélange carboglace-acétone) suivie d'hydrolyse, est une très bonne méthode de préparation des hydroperoxydes.
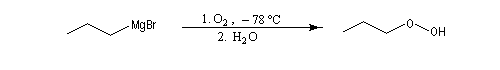
Avec le soufre, la réaction s'effectue par union directe entre le magnésien. Après hydrolyse, on obtient un thiol ou un thiophénol.
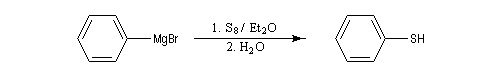
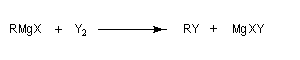
Réactions secondaires des organomagnésiens
Enolisation
Dans le cas de substrats encombrés, l'énolisation du substrat par l'organomagnésien jouant le rôle de base, peut devenir plus rapide que l'addition nucléophile sur le carbonyle.
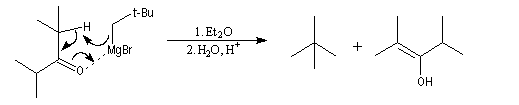
Réduction
Cette réaction intervient spécialement lorsque l'organomagnésien possède un atome d'hydrogène sur l'atome de carbone en b.
La réaction entre la 4,4-diméthylpentan-2-one et le bromure de 2-méthyléthylemagnésium suivie d'hydrolyse, fournit trois produits : A côté de l'alcool tertiaire (1) qui provient de l'addition de l'organomagnésien sur la cétone, on obtient un alcool secondaire (2) et du méthylpropène (3).
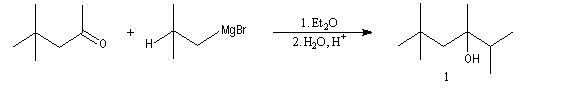
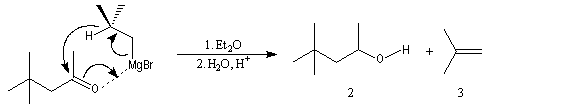
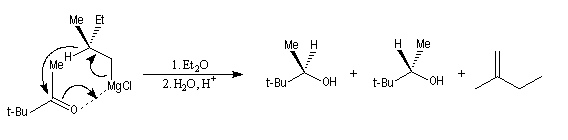
L'utilisation d'organomagnésiens chiraux a permis à H. S. Mosher en 1950, de réaliser la réduction énantiosélective de cétones encombrées [31].
La cétone de Michler (4,4'-bis(diméthylamino)- benzophénone) réagit avec les organomagnésiens pour donner des composés fortement colorés. Le complexe de transfert de charge initial fournit après hydrolyse un alcool. En milieu acide, ce dernier peut conduire à un carbocation tertiaire relativement stable.
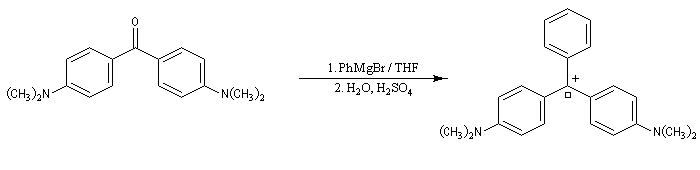
 |
L'erlenmeyer contient une solution de bromure de phénylmagnésium dans l'éthoxyéthane. On ajoute successivement 1 mL d'une solution de cétone de Michler dans le toluène, 1 mL d'eau distillée puis quelques gouttes d'acide acétique (éthanoïque). Après agitation, une couleur bleu vert de développe. Le carbocation obtenu est utilisé comme colorant. Celui-ci est appelé vert malachite en raison de la ressemblance de sa couleur avec celle de la malachite CuCO3(OH)2 : un minerai de cuivre (composé avec lequel il n'a bien entendu aucune parenté chimique). Porter des gants pour manipuler ce composé. |
- époxydes ;
- composés carbonylés [5], [6] ;
- carbonylés a, b-éthyléniques
- chlorures d'acyles ;
- esters ;
- orthoester [2] ;
- amides ;
- nitriles ;
Préparation
Les organolithiens les plus simples peuvent être obtenus par réaction entre le métal et un dérivé halogéné. Compte-tenu de la grande réactivité de ces composés il est indispensable d'utiliser des réactifs rigoureusement anhydres et de travailler en l'absence d'oxygène. On utilise un montage du même type que le précédent et on procède à un balayage soigneux du dispositif par un gaz inerte. L'utilisation d'une cuve à ultrasons permet d'accroitre la vitesse de la réaction en provoquant un décapage du métal. Différents solvants peuvent être utilisés parmi lesquels les éthers, le pentane, le cyclohexane.
Le butyllithium est un réactif très utile qu'on peut préparer par la réaction suivante :
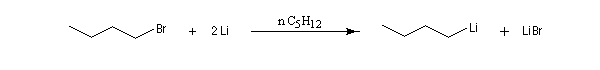 |
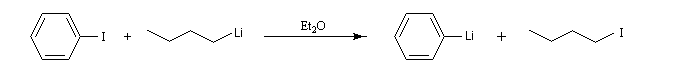
Puisque ce sont des carbanions potentiels, les organométalliques réagissent avec les composés possédant un atome d'hydrogène mobile. La position de l'équilibre acido-basique dépend de la différence des pKa des couples concernés.
Composé
|
n-BuLi
|
s-BuLi
|
t-BuLi
|
déprotone les hydrocarbures jusqu'à pKa
|
46
|
51
|
54
|
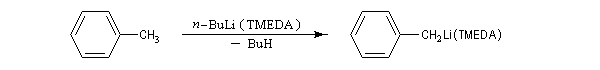
Des exemples d'utilisation du butyllithium en tant que base, concernent la préparation de carbanions stabilisés par des hétéroatomes :
- les ylures de phosphore utilisés dans la réaction de Wittig ;
- les ylures de soufre dans la réaction de Corey-Chaykowsky ;
- la métallation des dithianes de la réaction de Corey et Seebach.
- la préparation de carbanions silylés utilisés dans la réaction de Peterson.
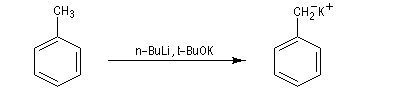
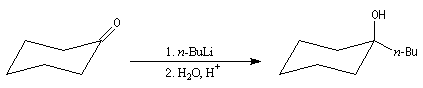
Pour utiliser les organolithiens dans des conditions optimales quand le solvant est l'éther, il est préférable d'effectuer leur dosage avant réaction car les lithiens attaquent lentement le solvant. L'organométallique provoque une réaction d'élimination en arrachant un d'hydrogène situé sur un atome de carbone en b de l'oxygène.
Déprotonation énantiosélective
La synthèse énantiosélective à partir d'organométalliques chiraux n'est possible que si l'on empêche l'équilibre d'inversion stéréochimique du centre chiral qui est un carbanion potentiel. Le complexe entre la (-)-spartéine, un alcaloïde extrait du lupin blanc, et un organolithien a été utilisé pour effectuer des réactions de déprotonation énantiosélectives. Un exemple est fourni par la préparation de dérivés de la pyrrolidine [10].
On commence par effectuer la protection de la fonction amine par le groupe t-butoxyxarbonyle.
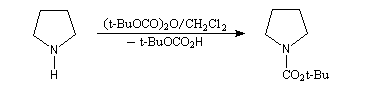
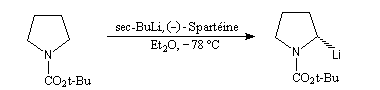
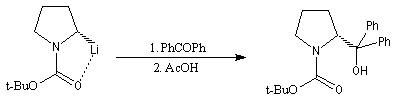
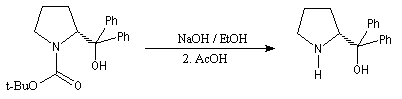
Les organolithiens sont suffisamment nucléophiles pour donner une réaction d'addition avec les ions carboxylates. Après hydrolyse, on obtient une cétone. Si le substrat est l'acide carboxylique, la réaction d'addition sur le carbonyle est précédée d'une réaction acido-basique qui consomme un équivalent de lithien. Il faut utiliser deux équivalents d'organolithien dans cette synthèse. La préparation de cétones à partir d'acides carboxyliques est étudiée dans le chapitre correspondant. Les organomagnésiens ne sont pas suffisamment nucléophiles pour donner cette réaction.
Préparation d'alcools encombrés
La synthèse des alcools encombrés peut être réalisée en utilisant des organolithiens. Ceux-ci étant beaucoup plus réactifs que les organomagnésiens, l'addition nucléophile peut être effectuée à basse température (- 70 °C) ce qui minimise les réactions d'énolisation et de réduction observées avec les organomagnésiens.
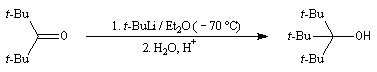
Préparation
Ces composés sont généralement préparés par transmétallation entre un organomagnésien et un sel de cadmium ou de zinc.
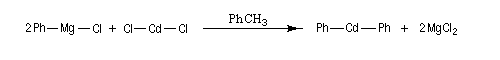
Organocuprates
Préparation des organocuprates
Les cuprates lithiens ont été introduits en synthèse organique par le chimiste américain H. Gilman (Iowa State University ) [29]. Il peuvent être préparés à partir d'un organolithien et d'une suspension d'iodure de cuivre (I) dans un solvant aprotique comme l'éthoxyéthane ou le THF à basse température sous atmosphère inerte. Ils forment des oligomères (R2CuLi)n.
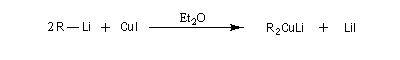
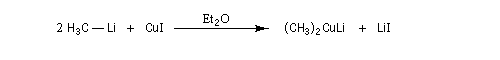
Les cuprates de Gilman permettent la préparation d'alcanes par couplage avec des dérivés halogénés saturés (réaction de Corey-House-Posner-Whitesides). Avec des substrats allyliques, on obtient des composés éthyléniques substitués.
A la suite des travaux de J. -F. Normant, il est possible de préparer par transmétallation des organocuprates portant un groupe vinyle à partir d'organomagnésiens vinyliques (réactifs de Normant.)
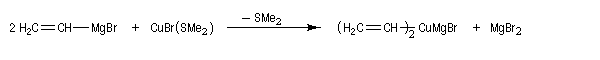
Les cuprates vinyliques peuvent être préparés par carbocupration entre un cuprate lithien et un alcyne. Ils permettent la préparation d'halogénures vinyliques de stéréochimie déterminée. Leur alkylation par dérivé halogéné permet la synthèse d'alcènes substitués avec une grande stéréosélectivité.
Cuprates zinciques A la différence des magnésiens ordinaires, les organozinciques sont compatibles avec une grande variété de groupes fonctionnels (GF) tels que : ester, nitrile, amine etc. Malheureusement ils sont peu nucléophiles. Leur emploi est donc assez limité.
L'addition d'une quantité catalytique de CuCN, 2 LiCl permet de passer au cuprate de Knochel beaucoup plus réactif qu'un organozincique ordinaire
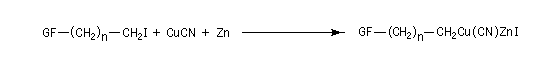
En ajoutant CuCN à une solution de cuprate de lithium, le chimiste américain Bruce Lipshutz a découvert une nouvelle famille de cuprates de formule générale : RnCu(CN)Lin. Avec : n = 1 (cuprates 1 : 1) ou n = 2 (cuprates 2 : 1). Dans ce dernier cas, ce sont des cuprates d'ordre supérieur (en anglais : higher order cuprates). Leur structure est encore l'objet de controverses. Leur réactivité est plus élevée que celle des cuprates de 1 : 1 [36].
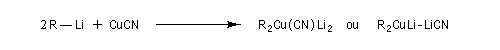
Tous ces cuprates réagissent de façon régiosélective en 1, 4 sur les composés carbonylés a, b-insaturés avec une grande stéréosélectivité.
Couplage d'Ullman
La réaction d'Ullman est une réaction de couplage des iodures (ou des bromures) de phényle en présence de cuivre, conduisant à des biaryles symétriques.
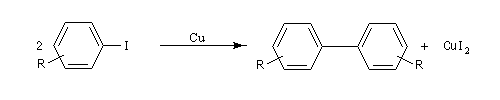
Du fait de sa limitation à la synthèse des composés symétriques et une température de travail assez élevée, on la remplace de plus en plus par des réactions de couplage croisé utilisant le palladium.
Organozinciques
Préparations
La préparation du premier organozincique, l'iodure d'éthylzinc, par Sir E. Frankland en 1848 marque le début de l'utilisation des composés organométalliques en synthèse organique. On écrivait à l'époque la réaction sous la forme.


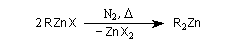
On peut aussi les préparer par réaction entre l'organomagnésien correspondant et ZnCl2 catalysée par le palladium. Les termes les plus simples comme le diéthylzinc sont des liquides dans les conditions ordinaires. Ils sont pyrophoriques et doivent être manipulés sous atmosphère inerte (Ar ou N2) [16]. Structure des zinciques
Contrairement aux réactifs RLi et RMgBr, les dérivés de type R2Zn, ne sont pas naturellement réactifs. Leur structure linéaire les rend non polaires et donc inertes. L'ajout d'un ligand permet de donner à l'organométallique chélaté une structure tétraédrique réactive.
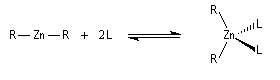
Le zinc étant fortement réducteur, la réaction entre ce métal et un dérivé halogéné peut conduire à une réaction de réduction. Ainsi la réaction entre le zinc et le chlorure de triphénylméthyle conduit à la formation d'un radical organique relativement stable, le radical triphénylméthyle.
Les organozinciques réagissent avec les dérivés halogénés en présence d'un catalyseur au palladium pour conduire à la réaction de couplage de Negishi.
Cyclopropanation
La cyclopropanation des composés éthyléniques peut être effectuée au moyen de la réaction de Simmons-Smith. Le réactif est préparé à partir d'un mélange de CH2I2 et du couple Zn-Cu. La solution est le siège d'un équilibre du type équilibre de Schlenk.
Le mode opératoire de la synthèse du bicyclo[4,1,0]heptane (norcarane) est donné à la référence [14]. Une autre méthode consiste a effectuer l'addition entre une double liaison éthylénique et un dichlorocarbène puis a effectuer une réduction du dihalogénure obtenu.
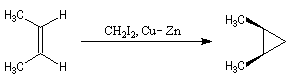
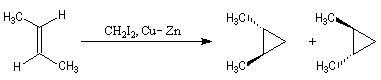
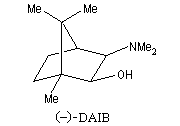
Addition énantiosélective des dialkylzinciques
L'addition des dialkylzinciques sur un aldéhyde possédant un carbone prochiral peut être énantiosélective en présence d'un auxilliaire chiral approprié. L'exemple ci-dessous concerne l'addition de diéthylzinc en présence de (-)-3-exo-(diméthylamino)isobornéol ou (-)-DAIB, par R. Noyori [32]. Le mode opératoire est donné à la référence [13].
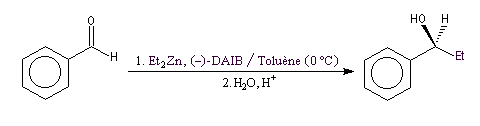
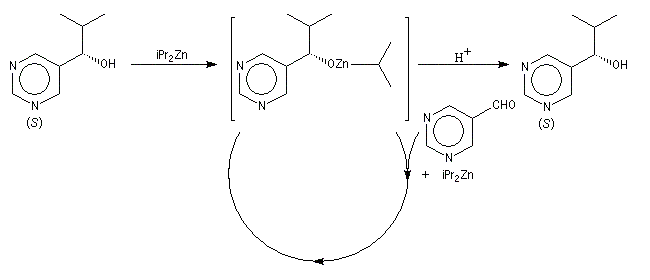
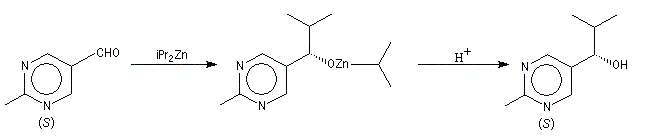
Considérons deux atomes A et B liés entre-eux par une liaison s et susceptibles de se lier à un complexe MLn selon le schéma suivant :
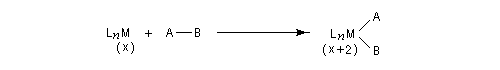
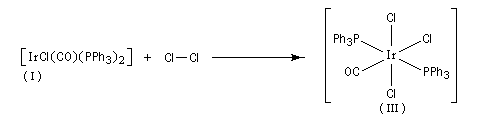
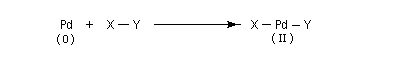
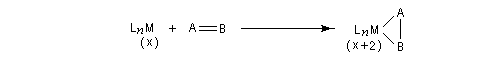
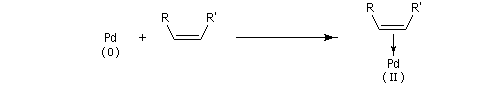
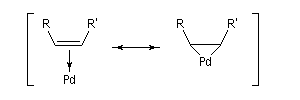
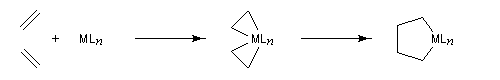
- mécanisme à trois centres ;
- mécanisme par substitution SN2 ;
- mécanisme ionique ;
- mécanisme radicalaire.
Au cours d'une élimination réductrice, le nombre de coordination du complexe ainsi que le nombre d'oxydation du métal décroissent. Ce processus peut être résumé par l'équation suivante :
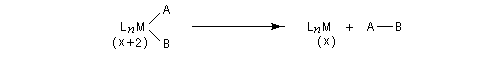
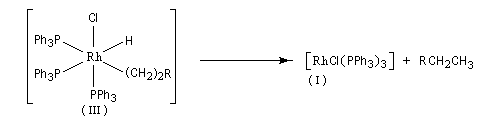
Il s'agit d'une réaction d'échange d'élément métallique.
Composés organopalladés Généralités
Les réactions organiques mettant en jeu le palladium peuvent être schématiquement réparties en deux catégories :
- celles qui font intervenir le Pd (II). Ce sont principalement des oxydations telles que celles mise en jeu dans l'oxydation des composés éthyléniques par le procédé Wäcker. Des composés du Pd (II) sont également utilisés comme catalyseurs dans d'autres réactions. Les composés du Pd (II) les plus utilisés sont PdCl2 et Pd(OAc). Ils sont stables et disponibles dans le commerce.
- les complexes du Pd (0) sont utilisés comme catalyseurs dans
plusieurs réactions organiques importantes. Notamment les réactions de
couplage permettant la création de nouvelles liaisons carbone-carbone.
Point important à noter : Pd est un métal noble. Aussi le Pd (0) est
plus stable que le Pd (II).
Les composés du Pd (II) les plus utilisés sont PdCl2, Pd(OAc). Ce dernier présente l'avantage par rapport au précédent d'être soluble en milieu organique.
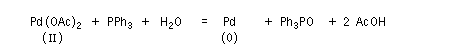
Couplages croisés utilisant le palladium
Le Pd (0) est utilisé comme catalyseur dans des réactions de couplage croisé. Bibliographie
F. A. Carey, R. J. Sunberg - Advanced organic chemistry (Plenum press 1996).
J. March - Advanced Organic Chemistry (J. Wiley 2006).
P. Laszlo - Logique de la synthèse organique - Cours de l'Ecole polytechnique (Ellipses 1993).
Les complexes de palladium en synthèse organique : initiation et guide pratique, J.-M. Campagne, D. Prim, Ed. Lavoisier, 2001.
N. N. Greenwood, A. Earnshaw - Chemistry of the elements (Pergamon Press 1986).
Metal-catalyzed Cross-coupling Reactions, Edited by F. Diederich and P. J. Stang, Wiley-VCH, New York, 2004
Organometallics in Synthesis. A manual, Schlosser, M., Second Edition, Wiley, New York, 2002.
Liens
[1] The Nobel Prize in Chemistry 1912
[2] n-Hexaldéhyde by G. Bryant Bachman.
[3] Highly reactive magnesium by R. D. Rieke, S. E. Bales, P. M. Hudnall, T. P. Burns, and G. S. Poindexter.
[4] Cyclohexylcarbinol by Henry Gilman and W. E. Catlin.
[5] Triphenylcarbinol by W. E. Bachmann and H. P. Hetzner.
[6] Manganese copper conjugate addition of Grignard Reagent by S. Marquais, M. Alami, and G. Cahiez.
[7] (Z)-1-iodohexene by A. Alexakis, G. Cahiez, and J. F. Normant.
[8] Di n-butyldivinyletain by Dietmar Seyferth.
[9] Enantioselective addition of butyllithium by D. Seebach and A. Hidber.
[10] (R)-(+)-2-(hydroxymethyl)-pyrrolidine by Nikola A. Nikolic and Peter Beak.
[11] Diethyl-Zinc by C. R. Noller
[12] Copper-Catalysed Conjugated Addition of Functionalized Organozinc Reagents to a, b-Unsaturated Ketones by B. H. Lipshutz, M. R. Wood, and R. Tirado
[13] Catalytic Enantioselective Additions of Dialkyl Zincs to Aldehydes using (2S)-DAIB) by Masato Kitamura, Hiromasa Oka, Seiji Suga, and Ryoji Noyori.
[14] Norcarane by R. D. Smith and H. E. Simmons
[15] Thèse Delacroix
[16] Manipulations sous atmosphère inerte modes opératoires pour la préparation des réactifs organométalliques
[17] Organozinc
[18] Cours P. Sibi.
[19] Structure des cuprates.
[20] New Methods for the Synthesis of Organozinc ans Organocopper Reagents by C. Piazza.
[21] Recent Advances in Organocurate Chemistry Jason S. Tedrow Evans Group, Harvard University.
[22] Réaction de Réformatsky asymétrique par Michel Obringer.
[23] Kumada coupling
[24] Organometallic Hypertext Book by Rob Toreki
[25] La chimie des organomagnésiens en France après Grignard par H. Normant.
Articles
[29] H. Gilman, R. G. Jones and L. A. Woods, J. Org. Chem., 1952, 17, 1630–1634.
[30] Watson, S. C.; Eastham, J. F. J. Organometal. Chem. 1967, 9, 165.
[31] D. O. Cowan, H. S. Mosher, Comparison of the Reactions of Grignard Reagents and Dialkylmagnesium Compounds in Addition, Reduction, and Enolization Reactions - J. Org. Chem., 27, p. 1-5.
[32] R. Noyori et al J. Am. Chem. Soc, 108, 6071, 1986.
[33] K. Soai. Nature 1295, 378, 767.
[34] K. Soai. Tetrahedron : Asymmetry, 1997, 8, 1717.
[35] Kohei Tamao, Koji Sumitani, Makoto Kumada (1972). "Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes". J. Am. Chem. Soc. 94 (12): 4374–4376. doi:10.1021/ja00767a075.
[36] B.H. Lipschutz, R.S. Wilhelm et D.M. Floyd, J. Am. Chem. Soc., 103 (1981) 7672.
[37] Kochi, J., Synthesis 1971, 303.
[38] J. -P. Foulon - Dosage des organomagnésiens et des lithiens. Bulletin de l'union des physiciens 629, décembre 1980.
[39] G. Dupuis - La découverte du radical triphénylméthyle par Moses Gomberg. Bulletin de l'Union des Physiciens, janvier 2002.
Pour aller plus loin
Synthèse organique par voie organométallique par T. Ollevier
Organometallic compound of Lithium Sodium and Potassium by Michael K. Denk
Organometallic chemistry by Worawan Bhanthumnavin
The basic chemistry of organopalladium compounds









0 commentaires:
Enregistrer un commentaire