Cours de chimie Organique -
Systèmes conjugués
Introduction
Définitions
Il y a plusieurs définitions possibles d'un système conjugué. Nous utiliserons la suivante : on appelle système conjugué, un ensemble d'atomes ayant des orbitales atomiques pouvant se recouvrir latéralement et permettant de fortes interactions électroniques entre au moins trois atomes contigus [15], [17]. Les systèmes conjugués ont une réactivité originale car la délocalisation entraîne une certaine ambiguïté dans la répartition électronique. Dans les exemples suivants, le système est représenté par des formes mésomères limites :
- systèmes p s p : à cette catégorie appartiennent les diènes conjugués dont le représentant le plus simple est le buta-1, 3-diène ;
les a-énones sont aussi des systèmes de ce type.
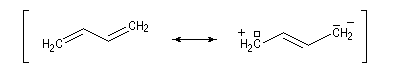
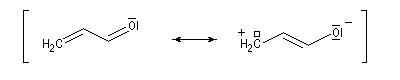
- systèmes n s p : dans cette catégorie, on trouve par exemple les énols et les énolates de composés carbonylés.
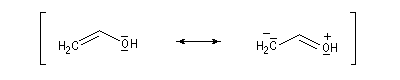
- systèmes p s v : comme exemple de tels systèmes nous prendrons celui du cation prop-2-ényle appelé de façon courante cation allyle.
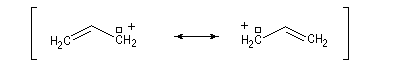
- systèmes n s v : le recouvrement des orbitales ne concerne que deux atomes, on ne peut plus vraiment parler de systèmes conjugués.
La délocalisation électronique explique la plus grande stabilité des carbocations possédant un hétéroatome en a par rapport aux carbocations ordinaires. Tout hétéroatome possédant un doublet non liant décrit par une orbitale de géométrie adéquate peut convenir. Les ions acylium en constituent un exemple :
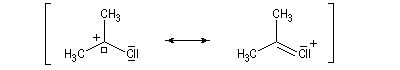
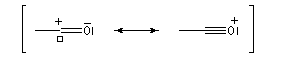
 |
 |
 |
|
non conjugué
|
conjugué
|
non conjugué
|
Les hydrocarbures aromatiques sont des polyènes conjugués cycliques ou [p]-annulènes répondant à des critères particuliers. Ils font l'objet d'un chapitre spécifique.
Quelques diènes conjugués importants
 |
L'isoprène est le 2-méthylbuta-1, 3-diène. Il constitue le motif de base formel des terpènes. La polymérisation stéréocontrôlée de l'isoprène permet la préparation du polyisoprène de stéréochimie cis dont la structure est la même que celle du caoutchouc naturel |
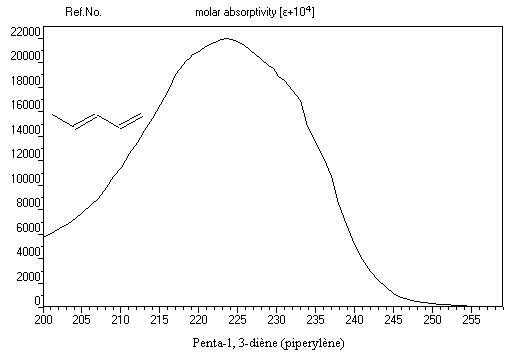
Domaine ultraviolet-visible
Le spectre ci-dessous est celui du penta-1, 3-diène. Il est typique d'un spectre de diène. On observe une bande large dont le maximum d'absorption est voisin de 225 nm.
|
Composés
|
lM (nm)
|
log e
|
|
éthène
|
162,5
|
3,94
|
|
buta-1, 3-diène
|
217
|
4,32
|
|
hexa-1, 3, 5-triène
|
247
|
4,53
|
Plus la différence d'énergie est grande, plus la longueur d'onde du maximum d'absorption est faible et plus le nombre d'onde de cette transition est élevé. La méthode de Hückel dans sa version la plus simple permet de rendre compte de ce phénomène. Il existe une corrélation très nette entre le nombre d'onde du maximum d'absorption mesuré et la différence d'énergie entre la plus haute orbitale moléculaire occupée (HO) et la plus basse vacante (BV) calculées selon la méthode de Hückel, exprimée ici en unités b.
Ces règles permettent la prévision de la longueur d'onde du maximum d'absorption des diènes conjugués. Le tableau ci-dessous donne quelques valeurs caractéristiques dans le solvant éthanol mais elles sont pratiquement insensibles à l'influence du solvant.
Pour calculer une longueur d'onde au moyen de cette méthode, on commence par considérer une structure de base :
|
Structure de base
|
|
 |
|
l de base (nm)
|
214
|
253
|
|
Substituant
|
Quantité à ajouter par substituant (nm)
|
|
double liaison conjuguée double liaison exocyclique alkyle, reste de cycle -NR1R2 -RO -Cl, -Br |
30
5 5 60 6 5 |
|
base 4 groupes alkyles : a, b, g, d 2 doubles liaisons exocycliques |
253 nm
20 nm 10 nm |
| lM (calculée) |
283 nm
|
La conjugaison, par le relâchement de la force des liaisons multiples qu'elle entraîne, se traduit par une diminution des nombres d'onde des absorptions des principaux vibrateurs de l'ordre de 20 cm-1.
Introduction
Les composés contenant le groupe prop-2-ényle sont appelés traditionnellement composés allyliques.
 |
Le 1-bromo-prop-2-ényle ou bromure d'allyle peut être préparé par bromation allylique du propène par le N-bromosuccinimide (NBS). |
Cations allyliques
Lorsqu'on effectue la solvolyse du 3-chlorobut-1-ène ou chlorure d'a-méthylallyle par un mélange eau-dioxane, on observe que la réaction est du premier ordre par rapport à l'halogénure. On obtient deux alcools I et II. I est le produit de substitution normalement attendu dans ce type de réaction tandis que II possède une structure dans laquelle le squelette carboné a subi une transposition.
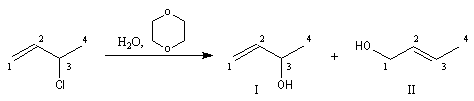
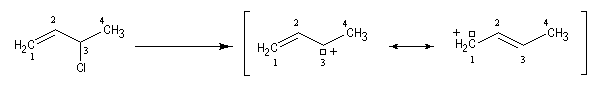
- le premier est le produit d'addition normal ;
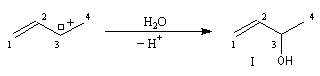
- le second correspond à un produit dans lequel la chaîne carbonée a été modifiée. La substitution s'est accompagnée d'un réarrangement des liaisons. C'est un exemple d'une réaction générale : la transposition allylique.
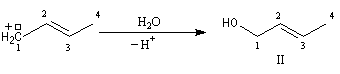
Mécanisme SN2'
Si l'on traite le 3-chlorobut-1-ène ou chlorure de a-méthylallyle par la triéthylamine, on constate que la réaction possède un ordre global égal à deux : premier ordre par rapport à la triéthylamine et premier ordre par rapport au substrat. Cette loi cinétique ne peut pas s'expliquer par un mécanisme SN1. On obtient 15 % de I et environ 85 % de II. On interprète ces résultats par le fait que I est le produit de substitution SN2 normal (substitution sur le C2) tandis que II correspond à une réaction du nucléophile sur le C4 qui implique une transposition.
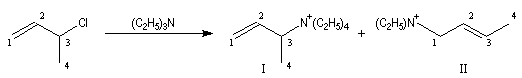
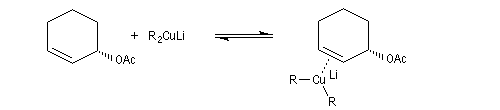
On observe une diminution de 10 unités de pKa pour le couple acido-basique quand on passe du propane (pKa = 50) au propène (pKa = 40). Autrement dit, le propène est environ dix milliards de fois plus acide que le propane.
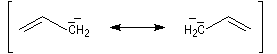
L'originalité de la structure allylique se manifeste également dans certaines réactions radicalaires. Le N-bromosuccinimide est un réactif permettant la bromation en a de la double liaison des composés éthyléniques. Il libère de très faibles quantités de brome, rendant ainsi négligeable la réaction concurrente d'addition. Cette réaction de bromation allylique est initiée par la lumière ou par des peroxydes servant d'initiateurs.
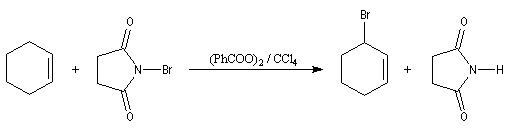
La méthode de Hückel simple (HMO) constitue une approximation de la théorie des orbitales moléculaires. Le principe de cette théorie a été rappelé dans un chapitre particulier.
Rappelons que les approximations conduisent à prendre :
- les intégrales coulombiennes Hmm égales pour des atomes identiques, par exemple deux atomes de carbone ;
- les intégrales de résonance (intégrales d'échange) Hmn égales entre atomes liés, nulles dans le cas contraire ;
- les intégrales de recouvrement Smn nulles entre atomes différents, tandis que Smm = 1 car les orbitales atomiques sont normalisées.
|
Hmm
|
Hmn
|
Hmn
|
Smn
|
|
a
|
b (atomes liés)
|
0 (atomes non liés)
|
dmn
|
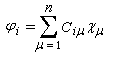
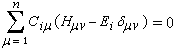
Passons à l'étude du système allylique. Sur la forme limite du schéma ci-dessous, l'atome 3 peut être affecté de 0, 1 ou 2 électrons. La résolution du système d'équations est la même pour tous les systèmes allyliques car la méthode HMO ne fait intervenir que la position des atomes.
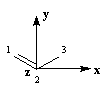 |
Les atomes de carbone et d'hydrogène sont situés dans un même plan. Le système
présente deux plans de symétrie :
|
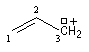 |
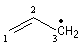 |
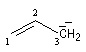 |
|
cation
|
radical
|
anion
|
On obtient un système de trois équations pour chaque valeur de l'énergie Ei :
(1) Ci1.xi + Ci2 = 0
(2) Ci1 + Ci2.xi + Ci3 = 0
(3) Ci2 + Ci3.xi = 0
Le déterminant séculaire s'écrit :
(xi2 - 2).xi = 0
il y a trois solutions, qui correspondent à trois valeurs de l'énergie, repérées par : i = 1, i = 2, i = 3 : x1 = - 1,41 ; x2 = 0 ; x3 = 1,41
- x1 = -1,41 ; le système s'écrit :
- -1,41 C11 + C12 = 0
- C11 - 1,41 C12 + C13 = 0
- C12 - 1,41 C13 = 0
la condition de normalisation se traduit par :
C112 + C122 + C132 = 1 - -1,41 C11 + C12 = 0
- x2 = 0 ; le système se réduit à deux équations :
- C22 = 0
- C21 + C23 = 0
la condition de normalisation se traduit par :
C212 + C232 = 1 - C22 = 0
- x1 = 1,41 ; le système s'écrit :
- 1,41 C31 + C32 = 0
- C31 + 1,41 C32 + C33 = 0
- C32 + 1,41 C33 = 0
la condition de normalisation se traduit par :
C312 + C322 + C332 = 1 - 1,41 C31 + C32 = 0
d'où : C11 = C13 = 0,5 ; C12 = 0,71
d'où : C21 = -C23 = 0,71
d'où : C31 = C33 = 0,5 ; C32 = -0,71
|
Energie
|
Ci1
|
Ci2
|
Ci3
|
|
E3 = a - 1,41b
|
0,5
|
- 0,71
|
0,5
|
|
E2 = a
|
0,71
|
0
|
- 0,71
|
|
E1 = a + 1,41b
|
0,5
|
0,71
|
0,5
|
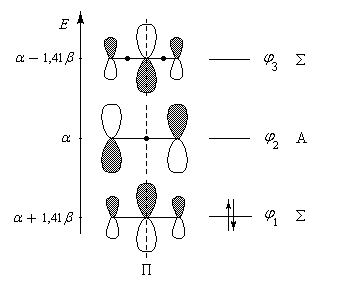 |
| Diagramme des orbitales moléculaires normalisées du système p du cation allyle. Les orbitales p sont symétriques (S) ou antisymétriques (A) par rapport au plan P. Par énergie croissante, ces fonctions possèdent respectivement 0, 1, 2 nœuds (point noir) |
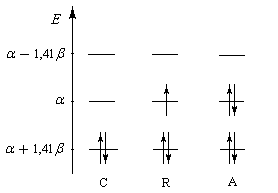 |
| Occupation des orbitales moléculaires p des systèmes allyliques : cation, radical, anion. Dans le cas du radical, la plus haute orbitale moléculaire occupée est appelée SOMO (single occupied molecular orbital). |
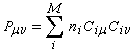
ni est le nombre d'électrons (0, 1, 2) décrits par l'orbitale moléculaire ji. La somme est étendue aux M orbitales occupées. Les résultats pour les systèmes précédents sont les suivants :
|
Indice de liaison
|
P12
|
P23
|
P13
|
|
cation
|
0,71
|
0,71
|
0,5
|
|
radical
|
0,71
|
0,71
|
0
|
|
anion
|
0,71
|
0,71
|
-0,5
|
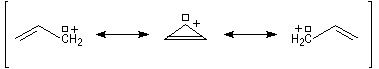
Résultats expérimentaux
Lorsqu'on s'intéresse aux distances interatomiques dans l'éthène, le buta-1, 3-diène et l'éthane, on obtient les résultats suivants :
|
Composé
|
Ethène
|
Buta-1,3-diène
|
Ethane
|
|
Distance interatomique
|
d(C1C2) = 0,134 nm
|
d (C2C3) = 0,148 nm
|
d (C1C2) = 0,154 nm
|
L'expérience montre que les conformations privilégiées de la molécule de buta-1, 3-diène sont celles pour lesquelles les atomes sont dans un même plan. Il existe deux conformations planes appelées s-trans (I) et s-cis(II).
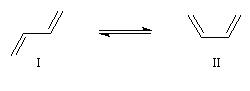
Ces résultats s'interprètent en utilisant la méthode de la mésomérie. La molécule peut-être considérée comme un hybride des formes limites suivantes :
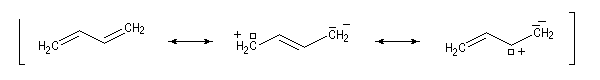
Nous allons maintenant utiliser une approche plus fine de la réalité grâce à la méthode des orbitales moléculaires. Méthode de Hückel appliquée au buta-1, 3-diène
Les principes de la méthode de Hückel ont été rappelés plus haut. La solution du problème est facilitée lorsqu'on considère les symétries de la molécule.
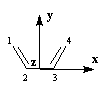 |
Les atomes de carbone et d'hydrogène sont situés dans un même plan. Dans la conformation s-cis, la molécule
présente deux plans de symétrie :
|
(1) Ci1.xi + Ci2 = 0
(2) Ci1 + Ci2.xi + Ci3 = 0
(3) Ci2 + Ci3.xi + Ci4 = 0
(4) Ci3 + Ci4.xi = 0
Les solutions de ce système reflètent la symétrie moléculaire. Elles sont symétriques ou bien antisymétriques par rapport au plan P. On peut, dès lors, les classer en deux groupes :
- solutions symétriques : Ci1 = Ci4 ; Ci2 = Ci3
le système d'équations s'écrit :
- Ci1.xi + Ci2 = 0
- Ci1 + Ci2.(xi + 1) = 0
dont le déterminant est : (xi + 1).xi- 1 = 0 ; xi2 + xi- 1 = 0 - Ci1.xi + Ci2 = 0
- solutions antisymétriques :
Ci1 = - Ci4 ; Ci2 = - Ci3
le système d'équations s'écrit :
- Ci1.xi + Ci2 = 0
- Ci1 + Ci2.(xi - 1) = 0
dont le déterminant est : (xi- 1).xi- 1 = 0 ; xi2-xi- 1 = 0 - Ci1.xi + Ci2 = 0
il y a deux solutions repérées par i = 1 et i = 3 : x1 = - 1,62 ; x3 = 0,62
il y a deux solutions repérées par i = 2 et i = 4 : x2 = - 0,62 ; x4 = 1,62
|
Energie
|
Ci1
|
Ci2
|
Ci3
|
Ci4
|
|
E4 = a - 1,62b
|
0,37
|
-0,60
|
0,60
|
-0,37
|
|
E3 = a - 0,62b
|
0,60
|
-0,37
|
-0,37
|
0,60
|
|
E2 = a + 0,62b
|
0,60
|
0,37
|
-0,37
|
-0,60
|
|
E1 = a + 1,62b
|
0,37
|
0,60
|
0,60
|
0,37
|
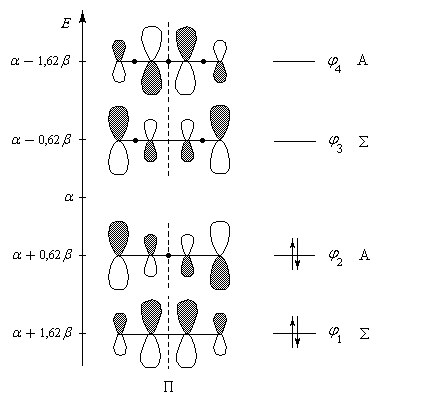 |
| Diagramme des orbitales moléculaires du système p du buta-1, 3-diène. Les orbitales p sont symétriques (S) ou bien antisymétriques (A) par rapport au plan P médiateur de la liaison C2-C3. Une autre caractéristique importante des fonctions d'onde est le nombre de nœuds (ou point nodal) qu'elle présentent, c'est à dire de points où la fonction s'annule en changeant de signe. Par énergie croissante, ces fonctions possèdent respectivement 0, 1, 2, 3 nœuds (point noir). On note que le nombre de nœuds croît de une unité quand on passe d'une om à la suivante (règle générale). |
La somme est étendue aux M orbitales occupées.
De nombreuses formules semi-empiriques ont été proposées pour relier la distance internucléaire Rmn à l'indice de liaison p : Pmn. En voici une :
Lorsque Pmn augmente, Rmn diminue. Pmn est un indicateur de la "force" de la liaison. A partir des coefficients des orbitales moléculaires calculés plus haut, on obtient les résultats suivants :
|
Liaisons
|
C1C2
|
C2C3
|
|
Pmn
|
0,89
|
0,45
|
|
Rmn (nm)
|
0,136
|
0,145
|
Energie de délocalisation électronique
Comme le montre le bilan énergétique de la réaction d'hydrogénation, un système conjugué comme le buta-1, 3-diène est plus stable qu'un système qui serait constitué de 2 doubles liaisons éthyléniques isolées. Pour chiffrer l'importance de la conjugaison sur la stabilité de la structure, on introduit la notion d'énergie de résonance électronique. C'est par définition, la différence entre l'énergie du système p et l'énergie de deux doubles liaisons non conjuguées modélisées par des molécules d'éthène :
Ed = Ep - EE
Ep = 2(a + 1,62b) + 2(a + 0,62b) ; Ep = 4a + 4,48b
EE = 2(a + b)
Ed = 0,48b
Ed < 0
Cela correspond bien à une stabilisation du système conjugué par rapport à un système constitué des liaisons non conjuguées. L'énergie de délocalisation électronique est une grandeur microscopique.
Energie de résonance
Enthalpie d'hydrogénation
L'hydrogénation catalytique du buta-1, 3-diène conduit au butane. Cette réaction n'a pas d'intérêt pratique mais elle est utile pour estimer le gain de stabilité dû à la conjugaison dans la molécule de buta-1, 3-diène par rapport à une structure non conjuguée.
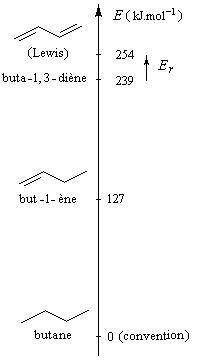 |
L'hydrogénation du buta-1, 3-diène dégage une énergie de -239 kJ.mol-1. Celle du but-1-ène dégage -127 kJ.mol-1 . On en déduit que l'hydrogénation du but-2-ène à deux doubles liaisons isolées fournirait :
2´ (-127) = -254 kJ.mol-1
L'énergie de résonance est la différence entre l'énergie de buta-1,
3-diène (hypothétique) et celle de la molécule de butadiène réelle.
C'est une grandeur positive par définition.
ER = 254 - 239
ER = 15 kJ.mol-1
Cette quantité est une
mesure de l'accroissement de stabilité qu'acquiert la molécule de
buta-1, 3-diène du fait de son caractère conjugué. |
la réaction d'addition du bromure d'hydrogène HBr sur le buta-1, 3-diène donne deux dérivés bromés isomères : le 3-bromobut-1-ène (1) et le 1-bromobut-2-ène (2). On les appelle traditionnellement produit d'addition 1, 2 et produit d'addition 1, 4.
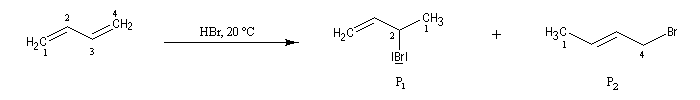
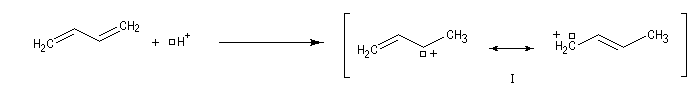
- l'addition de l'ion bromure peut s'effectuer sur l'atome de carbone 2 pour conduire au composé P1 ou produit d'addition 1, 2 ;
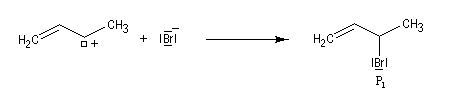
- elle peut aussi s'effectuer sur l'atome de carbone 4 pour donner le composé P2 ou produit d'addition 1, 4.
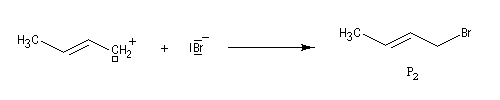
|
Température
|
0 °C
|
40 °C
|
|
P1
|
90 %
|
10 %
|
|
P2
|
15 %
|
85 %
|
- à la température de 0 °C, la réaction est sous contrôle cinétique. Le produit majoritaire est le plus rapidement formé. C'est P1 car l'état de transition qui y conduit est celui qui possède l'enthalpie libre molaire d'activation la plus basse ;
- à température plus élevée, la réaction passe sous contrôle thermodynamique. Les produits P1 et P2 sont alors en équilibre via le carbocation allylique I. Le produit majoritaire est alors le plus stable. C'est P2 car DrG 2* < DrG 1*.
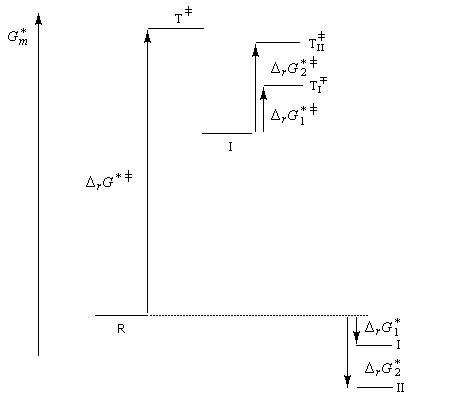
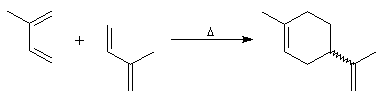
Introduction Lorsqu'on chauffe un mélange d'éthène et de butadiène à 200 °C, on obtient un mélange de cyclohexène et de 4-vinylcyclohexène. La transformation s'interprète par deux réactions :
- la première fournit du cyclohexène ;
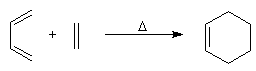
- la seconde conduit au 4-vinylcyclohexène qui est le produit majoritaire.
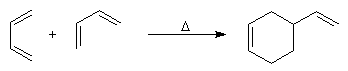
L'importance de la réaction de Diels-Alder vient du fait qu'elle est généralisable aux alcènes et aux diènes fonctionnalisés. On peut ainsi accéder aux cyclohexènes substitués qui constituent une partie du squelette de nombreuses molécules complexes. Un exemple est celui de plusieurs synthèses de la molécule de cantharidine pour lesquelles la réaction de Diels-Alder constitue l'étape clé. Cette réaction a fait l'objet d'un nombre considérable d'études tant d'un point de vue expérimental que théorique. La réaction de Diels-Alder a trouvé une interprétation particulièrement élégante dans la théorie de la conservation de la symétrie des orbitales développée par les chimistes américains R. B. Woodward et R. Hoffmann que ces derniers ont mise en évidence dans l'étude des réactions électrocycliques [19], [35] au cours de la synthèse de la vitamine B12 [34]. La réaction de Diels-Alder constitue aussi un bel exemple du rôle joué par les orbitales frontières des réactifs, théorie développée par le chimiste japonais K. Fukui. Ce dernier et R. Hoffmann se sont partagés le prix Nobel de chimie en 1981 [32]. Propriétés générales
La réaction est facilitée si l'un des partenaires (généralement le philodiène) porte un ou plusieurs groupes attracteurs d'électrons. Par exemple avec un nitroalcène comme diènophile.
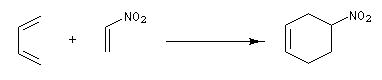
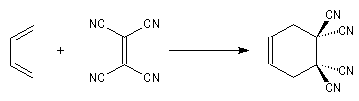
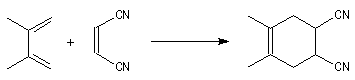
Les diènes cycliques comme le cyclopentadiène possèdent des doubles liaisons dans une configuration Z favorable et ils réagissent généralement plus rapidement que leurs homologues acycliques.
Le benzyne constitue un bon diénophile dans les réactions de Diels-Alder.
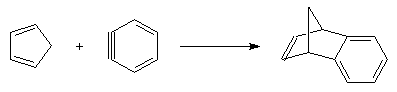
Les quinones sont de bons diénophiles dans les réactions de Diels-Alder et elles ont été utilisées dans les premières synthèses de stéroïdes.
La réaction peut être utilisée pour synthétiser certains hétérocycles. L'exemple suivant concerne une préparation du dihydropyrane (DHP) : un éther d'énol.
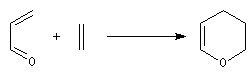

La réaction intermoléculaire est renversable. Elle est en effet exothermique (2 liaisons p sont remplacées par deux liaisons s) et elle a une entropie de réaction négative (état initial plus désordonné que l'état final). Ainsi, lorsqu'on élève la température, il est possible d'observer la réaction inverse au delà de la température d'inversion de l'équilibre. Celle-ci est appelée rétro Diels-Alder.
 |
Le cyclopentadiène peut être préparé par craquage
thermique du dicyclopentadiène à 150 °C. Cette réaction intervient dans
la synthèse du ferrocène [3].
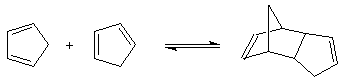 |

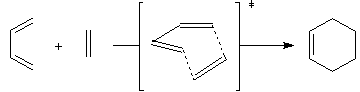
L'addition entre le butadiène et l'éthène ne peut pas s'interpréter par un contrôle de charge car les réactifs impliqués sont électriquement neutres. Par ailleurs, l'expérience montre que la réaction est concertée c'est à dire que des liaisons sont rompues et d'autres reformées sans qu'il y ait passage par un composé intermédiaire. Woodward et Hoffmann ont proposé d'appeler péricycliques les réactions concertées dans lesquelles les liaisons sont formées et rompues dans un état de transition cyclique [24]. La réaction de Diels-Alder appartient à cette catégorie.
Il s'agit ici plus précisément d'une cycloaddition mettant en jeu 4 électrons pour le premier réactif et 2 pour le second c'est pourquoi on l'appelle cycloaddition [4 + 2].
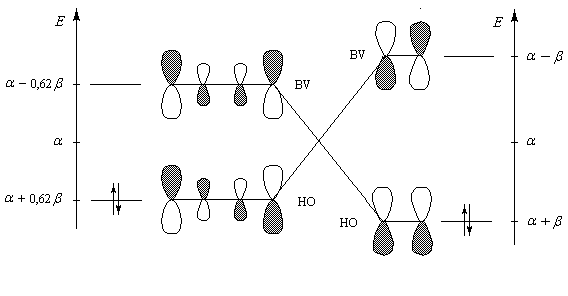 |
| Diagramme montrant l'interaction entre les orbitales frontières du butadiène et de l'éthylène dans la réaction de Diels-Alder. |
Expérimentalement, on constate que la constante de vitesse de la réaction croit lorsque l'un des partenaires est appauvri en électrons tandis que l'autre est au contraire enrichi en électrons. Le tableau ci-dessous regroupe quelques valeurs de la constante de vitesse pour la réaction suivante entre le cyclopentadiène et l'acrylonitrile.
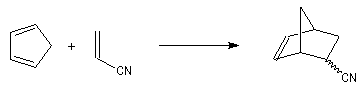
|
Diénophile
|
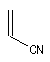 |
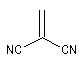 |
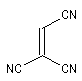 |
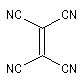 |
|
k
|
1
|
45,5´103
|
48,0´104
|
43,0´106
|
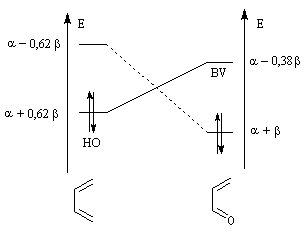
Examinons l'influence de la substitution sur les interactions entre HO et BV pour deux systèmes respectivement non substitué et substitué.
Dans les diagrammes ci-dessous, (I) est substitué par un groupe donneur d'électrons tandis que (II) est substitué par un groupe attracteur :
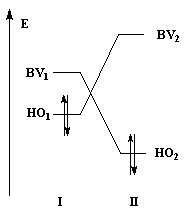 |
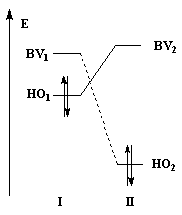 |
| Système non substitué : les différences entre les niveaux d'énergie des orbitales frontières des molécules (I) et (II) sont les mêmes. | Système substitué : l'énergie de la HO de (I) est relevée ; celle de la BV de (II) est abaissée. L'écart le plus faible entre les orbitales frontières met en jeu la HO de (I) et la BV de (II). |
 |
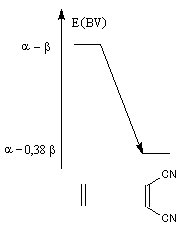 |
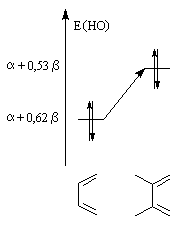
| Influence de la substitution du buta-1, 3-diène par deux groupes méthyle en 2 et 3 sur le niveau de la HO. | Influence de la substitution de l'éthène par deux groupes cyano sur le niveau de la BV. |
- la plupart du temps, l'interaction prédominante est celle
qui implique la HO du diène et la BV du diénophile. La réaction est dite
à demande électronique normale.
Dans l'exemple ci-dessous, les énergies de la HO et de la BV sont obtenus par la méthode de Hückel ;
- on connaît des exemples dans lesquels l'interaction
prédominante a lieu entre la HO du diénophile et la BV du diène. Il
s'agit de réactions à demande électronique inverse.
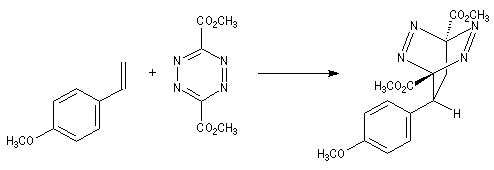
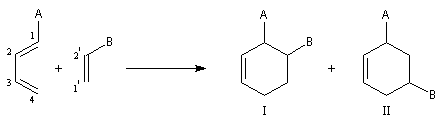
Lorsque le diène et le diénophile ne sont pas substitués de manière symétrique, deux adduits peuvent se former. Dans l'exemple ci-dessous les adduits (I) et (II) sont appelés respectivement ortho et méta par référence à la chimie des cycles benzéniques.
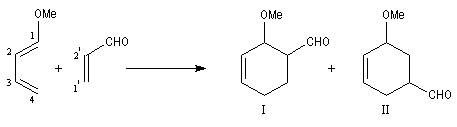
Lorsque le diène est substitué par un groupe donneur et le diénophile par un groupe attracteur, l'interaction prédominante fait intervenir la plus haute orbitale occupée du diène et la plus basse orbitale vacante du diénophile. La régiosélectivité peut être prévue en partant de l'idée que les liaisons s'effectuent entre les atomes de carbone possédant les coefficients les plus élevés dans ces orbitales.
|
Formules
|
 |
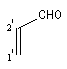 |
|
Composés
|
X
|
Y
|
|
Energie
|
C1
|
C2
|
C3
|
C4
|
|
EHO = a + 0,46b
|
0,50
|
0,47
|
-0,28
|
-0,60
|
|
EBV = a - 0,71b
|
0,61
|
-0,29
|
-0,41
|
0,58
|
|
Energie
|
C'1
|
C'2
|
|
EHO = a + b
|
-0,58
|
-0,58
|
|
EBV = a - 0,35b
|
-0,66
|
0,26
|
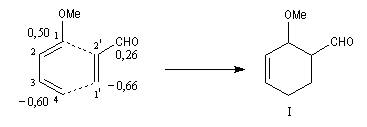
Le caractère syn de l'addition entre le diène et le diénophile entraîne la stéréospécificité. Celle-ci s'exprime doublement. Par rapport aux diène et par rapport au diénophile :
- stéréospécificité par rapport au diénophile ;
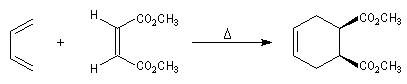
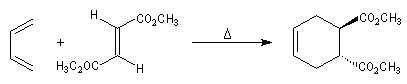
- stéréospécificité par rapport au diène ;
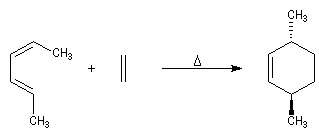
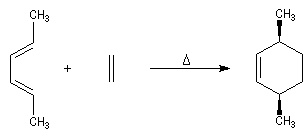
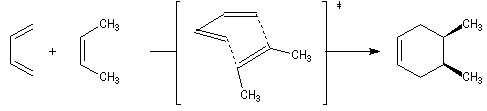
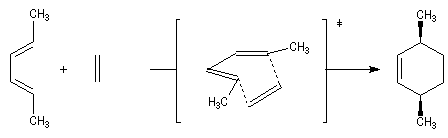
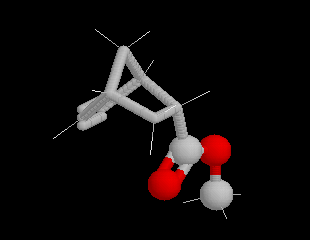
Lorsqu'on réalise une réaction de Diels-Alder entre un diène cyclique comme le cyclopentadiène et un dérivé substitué de l'éthène, deux adduits diastéréo-isomères I et II peuvent se former. Traditionnellement, on utilise une nomenclature propre aux composés bicycliques pontés. I est appelé composé endo tandis que II est appelé composé exo.
 |
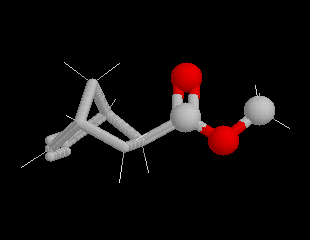 |
| Stéréoisomère endo : le groupe R est dirigé vers l'intérieur de la molécule, du côté opposé au pont le plus petit. | Stéréoisomère exo : le groupe R est dirigé vers l'extérieur de la molécule du même côté que le pont le plus petit. |
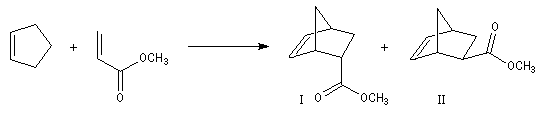
|
Composé
|
endo
|
exo
|
|
Pourcentage
|
78
|
22
|
- recouvrement principal : C1-C3' et C4-C2' ;
- recouvrement secondaire : C2-O et C3-C1'.
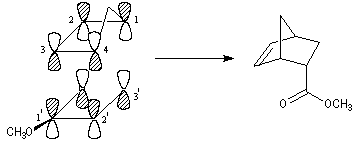
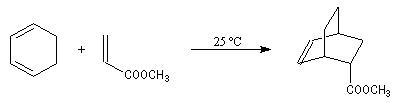
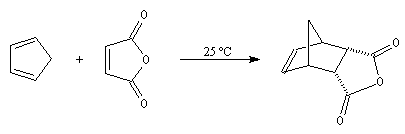
L'anhydride maléique est un excellent diénophile. L'adduit obtenu qui possède l'équivalent de deux fonctions acides est ensuite susceptible de fournir plusieurs produits intéressants.
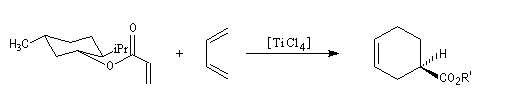
Le substrat est un ester du menthol. Ce dernier est utilisé comme auxilliaire chiral. L'acide de Lewis est le tétrachlorure de titane. Le produit est obtenu avec un excès énantiomérique de 90 %.
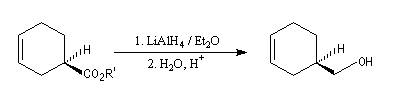
 |
Le patchoulol est un alcool de formule brute C15H26O. Il est issu de l’huile essentielle d’une famille de plantes, les patchoulis, voisines des menthes, et utilisé en parfumerie. L'oxyde de patchoulène qui peut être obtenu assez facilement à partir du patchoulol, a été utilisé comme composé de départ par R. A. Holton dans sa remarquable synthèse du taxol en 1994. |
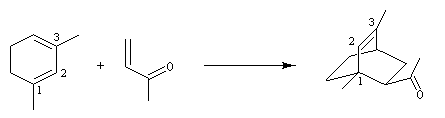
Synthèse du taxol
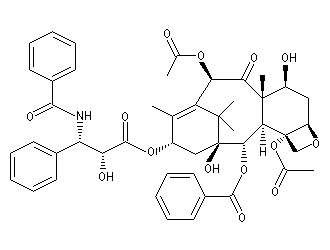 |
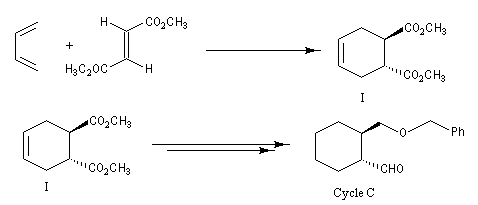
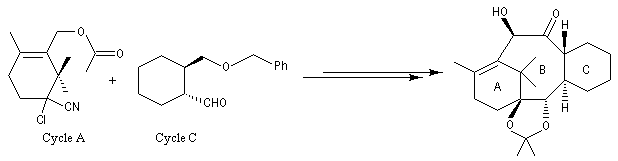
Le taxol (paclitaxel) est une substance extraite de l'écorce de l'If du pacifique (Taxus brevifolia), possédant des propriétés anticancéreuses. Il a été découvert en 1962 par le botaniste américain Arthur Barclay qui participait à un criblage de substances naturelles dans les forêts de l'Ouest américain dans le cadre d'un programme institué par le National Cancer Institute (NCI). La molécule a été isolée en 1971 à partir de l'écorce de l'If du pacifique par les chimistes Wall, Wani et Taylor. Il faut 1 kg d'écorce pour isoler 100 mg de produit actif. Une production à grande échelle à partir de cette seule source est donc difficile à envisager. C'est en essayant d'extraire des analogues du Taxol issus de l'if européeen que les chercheurs de l'Institut de chimie des substances naturelles de Gif sur Yvette on découvert le taxotère. La complexité de la molécule de taxol (la molécule possède 11 centres chiraux) a constitué un challenge pour de nombreuses équipes. Quelques étapes de la synthèse de K. C. Nicolaou sont décrites ci-dessous [62]. Signalons un autre médicament anticancéreux mis au point par la même équipe de l'ICSN : la Navelbine |
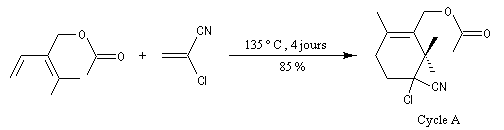
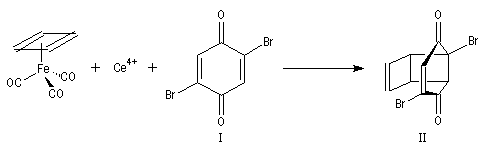
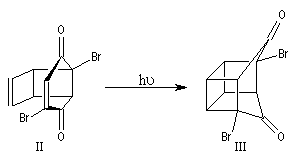
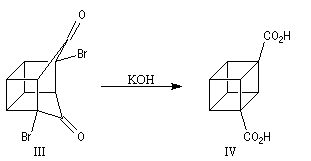
Cette synthèse, réalisée par J. C. Barborak, L. Watts et R. Petitt de l'Université du Texas en 1966, débute par une addition de Diels-Alder. Le diène est le cyclobutadiène provenant de la décomposition d'un complexe que Petitt avait préparé quelque temps auparavant. Le philodiène est un dérivé dibromé de la parabenzoquinone.

L'oxygène singulet joue le rôle de diénophile dans une réaction de cycloaddition [4 + 2] avec les diènes, apparentée à la réaction de Diels-Alder.
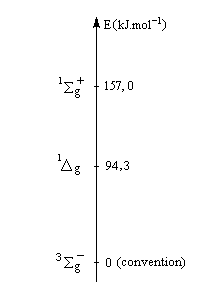 |
L'état fondamental de la molécule d'oxygène est un état triplet 3Sg. Les deux premiers états excités sont deux états singulets d'énergies respectives 94,3 et 157,0 kJ.mol-1 (schéma de gauche). L'oxygène singulet 1Dg peut être préparé par irradiation de O2 (3Sg) en présence d'un photosensibilisateur [47], [48]. 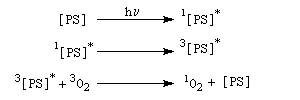
|
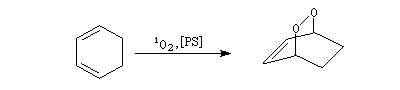
 |
La cycloaddition de l'oxygène singulet permet la synthèse
de l'ascaridole, un composé présent dans l'huile de chenopodium, à
partir de l'a-terpinène.
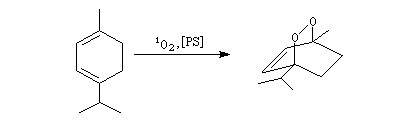 |
 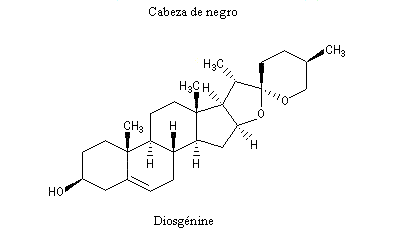 |
La progestérone, une hormone stéroïde, a été isolée en 1934. La première hémisynthèse efficace de la progestérone a été réalisée en 1941 par le chimiste américain Russel E. Marker (Université de Pennsylvanie) en utilisant un procédé de dégradation de la diosgénine, une molécule d'origine naturelle extraite des tubercules d'une dioscorée mexicaine (appelée : cabeza de negro). Russel E. Marker (Université de Pennsylvanie) fonda en 1944 le laboratoire pharmaceutique Syntex SA (contraction de synthèse et mexique) dans le but de commercialiser la progestérone obtenue par son procédé de dégradation de la diosgénine. Cette firme fait désormais partie du groupe chimique et pharmaceutique Helvétique Roche depuis 1994. |
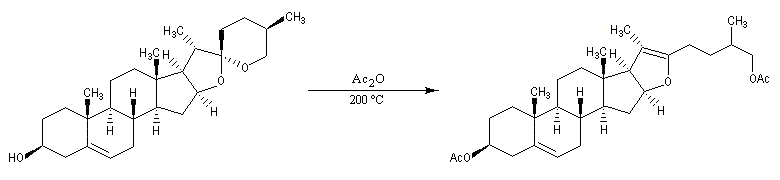

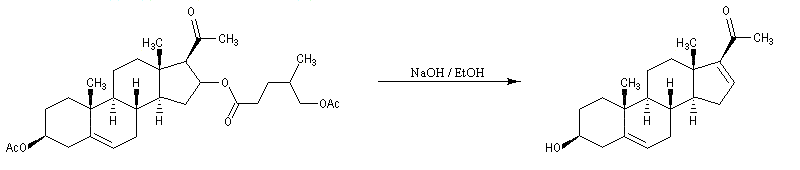
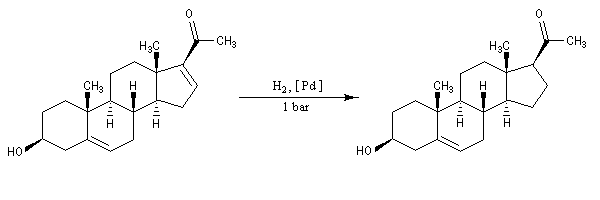
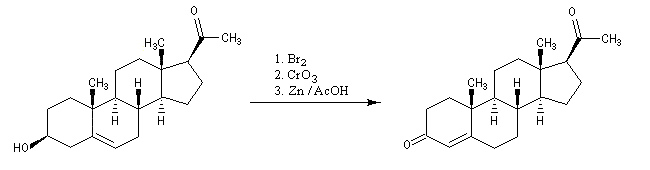
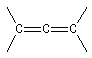
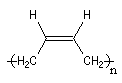
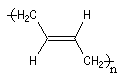
La polymérisation des diènes est, dans son principe, analogue à celle des autres composés éthyléniques. La présence de deux doubles liaisons offre des possibilités supplémentaires dans l'enchaînement des monomères. Ainsi, la polymérisation du buta-1, 3-diène peut fournir trois polymères différents :
- la polymérisation 1, 2 est celle d'un monomère vinylique ordinaire ;
- la polymérisation 1, 4 pose un problème stéréochimique du fait de la diastéréo-isomérie Z, E dont l'origine est la présence des doubles liaisons éthyléniques dans le polymère. Deux composés peuvent être obtenus :
 |
 |
|
Z
|
E
|
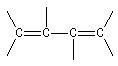
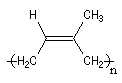
La polymérisation de l'isoprène est analogue dans son principe à celle du buta-1, 3-diène. A côté de la polymérisation 1, 2, on peut obtenir deux types de polymères 1, 4 :
 |
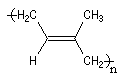 |
|
Z
|
E
|
Le caoutchouc naturel provient de la polymérisation naturelle stéréorégulière, catalysée par des enzymes, de l'isoprène. On trouve ce dernier dans le latex, produit par l'écorce de l'hévéa ou arbre à caoutchouc. Le caoutchouc naturel est le (Z)-polyisoprène-1, 4. Dans son beau livre les objets fragiles [23], P.-G de Gennes raconte comment la polymérisation naturelle de l'isoprène était mise à profit par les Indiens d'Amazonie pour se confectionner des bottes sur mesure.
Comme la molécule obtenue possède encore des liaisons doubles, il est possible de relier les chaînes entre-elles par des ponts. La formation de tels ponts intermoléculaires en présence d'une petite quantité de soufre s'appelle vulcanisation. Ce procédé, qui intervient dans la fabrication de pneumatiques, a été découvert par l'inventeur américain Charles Goodyear en 1839. Les liaisons pontales diminuent le comportement plastique et augmentent le caractère élastique. Dans les pneumatiques pour automobiles, on ajoute du carbone au caoutchouc vulcanisé pour augmenter la résistance de la gomme à l'abrasion.
Si la quantité de soufre est plus importante, on obtient un composé de couleur noire qui ne possède plus aucune propriété élastique, l'ébonite, dont la couleur rappelle celle de l'ébène et dont sont constituées les boules de bowling.
La polymérisation de l'isoprène peut être réalisée en utilisant des catalyseurs de Ziegler et Natta [43]. Elle conduit au (Z)-polyisoprène-1, 4 dont la structure est identique à celle du caoutchouc naturel. Le (E)-polyisoprène-1, 4 appelé gutta-percha, ne possède pas de propriétés élastiques. Il intervient dans la fabrication des balles de golf. Polymères conjugués Polyacétylène
La synthèse du polyacétylène a été vue dans le chapitre consacré aux alcynes. La structure du polyacétylène mérite quelques commentaires. On peut décrire la molécule par l'alternance de liaisons simples et de liaisons doubles. Ces liaisons ont des longueurs inégales. Les liaisons doubles sont plus courtes que les liaisons simples. Cette structure particulière, si on la compare à celle où les liaisons auraient une longueur égale, est la conséquence d'une distorsion comparable à l'effet pseudo Jahn-Teller décrit à propos du cyclobutadiène. Elle porte le nom de distorsion de Peierls. Dans la méthode de Hückel, on considère deux intégrales de recouvrement :
- b1 associée aux liaisons longues (simples) ;
- b2 associée aux liaisons plus courtes (doubles).
- la bande de valence est pleine avec un niveau de Fermi de valeur : a + (b2 - b1) ;
- la bande de conduction est vide.
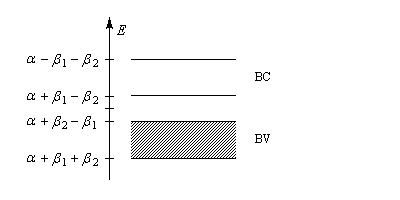
Il est possible de faire apparaître des charges électriques dans la structure du polyacétylène. Cette création de charge s'appelle dopage. Les premiers exemples de dopage de polymères comme le polyacétylène ont mis à profit des réactions d'oxydoréduction :
- par oxydation on obtient un dopage de type p
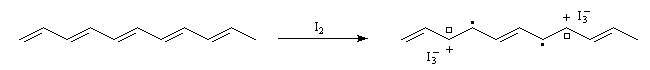
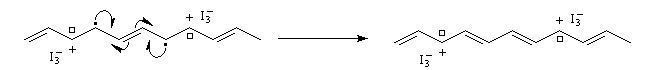
- par réduction au moyen d'un réducteur puissant comme le radical-anion naphtalène-sodium, on obtient un dopage de type n.
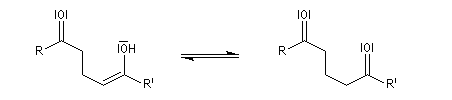
a-énones
Les a-énones sont des substrats intéressants en synthèse organique car ils donnent lieu à des réactions variées. On les obtient par plusieurs voies. Citons en quatre :
- oxydation des alcools allyliques (y compris les alcools tertiaires, avec transposition du squelette) ;
- déshydratation des aldols (et des cétols) ;
- réaction de Wittig-Horner ;
- réaction d'addition d'ions acylium issus de chlorures d'acyles sur les double liaisons éthyléniques suivie d'élimination.
 |
Le 3-hydroxybutanal ou aldéhyde crotonique est obtenu par déshydratation de l'aldol de l'éthanal.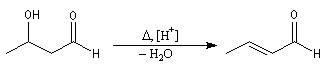 C'est un composé lacrymogène et toxique. Par hydrogénation ménagée il fournit le butanal. L'hydrogénation plus poussée fournit le butan-1-ol. |
 |
Le 4-méthylpent-3-éne-2-one ou oxyde de mésityle est obtenu facilement par déshydratation du cétol
de la propanone en milieu acide ou en présence d'une trace de diiode.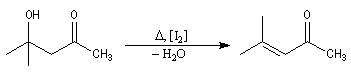 L'hydrogénation de l'oxyde de mésityle conduit à la méthylisobutylcétone, utilisée comme solvant des peintures et vernis. |
 |
Le propénal ou acroléine est obtenu industriellement par oxydation ménagée du propène
en présence de catalyseurs au molybdène ou en présence d'oxyde de cuivre (I). Il possède une odeur âcre. Il se forme par décomposition des graisses (oléines) d'où son nom : acroléine. Il intervient dans plusieurs synthèses industrielles comme celle des amino-acides D et L méthionine. Il est également utilisé dans la préparation du glycérol. |
 |
La jasmone est une a-énone contenant un cycle pentagonal et une chaîne latérale de stéréochimie Z. On la trouve à l'état naturel dans le jasmin.
La molécule de jasmone possède la structure ci-dessous :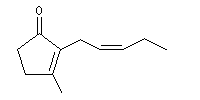
|
Structure électronique
Les a-énones sont des composés conjugués qui possèdent deux conformations remarquables. En raisonnant par analogie avec le cas du buta-1, 3-diène, on peut prévoir que la conformation privilégiée du propénal est la forme s-trans.
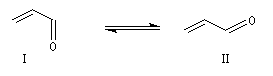
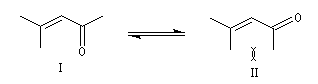
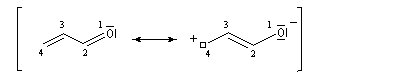
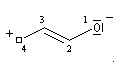
|
Energie
|
Ci1
|
Ci2
|
Ci3
|
Ci4
|
|
E4 = a - 1,55b
|
0,25
|
-0,60
|
0,65
|
-0,42
|
|
E3 = a - 0,38b
|
0,44
|
-0,56
|
-0,25
|
0,66
|
|
E2 = a + 0,99b
|
-0,58
|
-0,3
|
0,48
|
0,58
|
|
E1 = a + 1,91b
|
0,66
|
0,58
|
0,42
|
0,22
|
La charge électronique qm de l'atome m, est définie par :
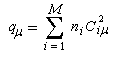
ni est le nombre d'électrons (0, 1, 2) qui sont décrits par l'orbitale moléculaire ji. La somme est étendue aux M orbitales moléculaires occupées.
La charge nette de l'atome m est la différence entre le nombre mm d'électrons apportés au système par cet atome et sa charge électronique :
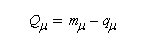
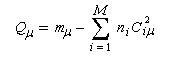
|
Q1
|
Q2
|
Q3
|
Q4
|
|
-0,54
|
0,33
|
-0,03
|
0,23
|
- un nucléophile dur, réagira sous contrôle de charge sur l'atome de carbone 2 qui développe la plus grande densité électronique ;
- un nucléophile mou, réagira de préférence sous contrôle orbitalaire sur l'atome qui possède le plus gros coefficient dans la BV c'est à dire sur l'atome de carbone 4.
Les transitions en ultraviolet des composés carbonylés non conjugués ont été évoquées dans le chapitre correspondant. Le spectre du 4-méthylpent-3-én-2-one ou oxyde de mésityle (solvant éthanol) est représenté ci-dessous :
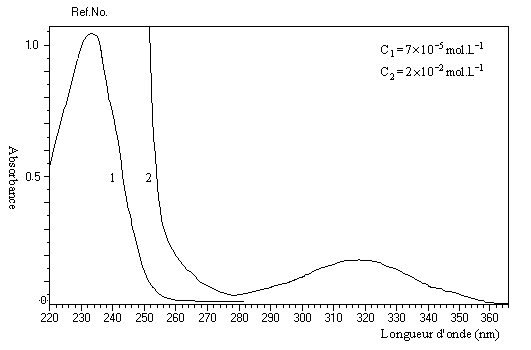
|
Composés
|
transition p ® p*
|
transition n ® p*
|
|
propanone
|
188 nm
|
279 nm
|
|
oxyde de mésityle
|
236 nm
|
315 nm
|
Les règles de Woodward permettent la prévision de la longueur d'onde du maximum d'absorption de la transition p ® p*. Les incréments à ajouter à la longueur d'onde de base dépendent de la position des substituants sur la structure et de sa nature. Les positions sont représentées par les lettres a, b, g, d :
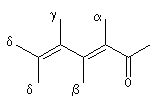
|
structure de base
|
 |
 |
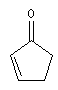 |
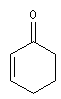 |
|
l de base (nm)
|
207
|
215
|
202
|
215
|
|
substituant
|
quantité à ajouter par substituant (nm)
|
|
double liaison exocyclique conjugaison supplémentaire diène homoannulaire |
5
30 39 |
|
position des substituants
|
a
|
b
|
g
|
d
|
|
-R, -RO, -CH3CO2,
-PhCO2 |
10
|
12
|
18
|
18
|
|
-OH
|
35
|
30
|
-
|
50
|
|
-Br
|
25
|
30
|
-
|
-
|
à la valeur de base de 215 nm, il faut ajouter 24 nm pour les groupes alkyles en a et b, soit un total de 239 nm en bon accord avec la valeur expérimentale.
L'exemple ci-dessous concerne la pulégone (du latin pulex : puce, en raison de la réputation de ce composé à éloigner ces insectes).
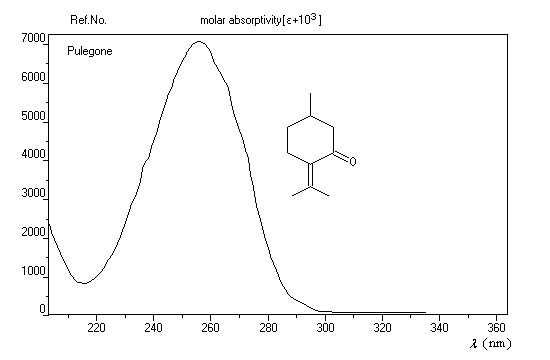
Les absorptions caractéristiques des aldéhydes et des cétones en spectroscopie infrarouge ont été étudiées dans le chapitre sur les composés carbonylés.
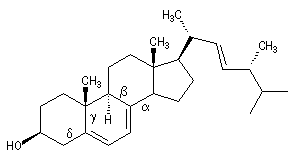
Du fait de leur caractère conjugué, les a-énones sont plus stables que les b-énones. Lorsque les conditions sont requises, celles-ci ont tendance à conduire aux premières par isomérisation. La migration de la double liaison est catalysée par les acides. L'exemple suivant concerne l'isomérisation de la cholest-5-éne-3-one (I) en cholest-4-éne-3-one (II) plus stable.
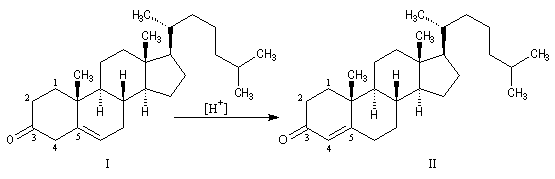
La préparation des réactifs est décrite dans le chapitre sur les composés organométalliques. Les principaux résultats sont donnés ci-dessous :
- les organolithiens fournissent préférentiellement le produit d'addition 1, 2. Le cation lithium, petit et peu polarisable est un acide dur, (h = 35,1 eV). Son association avec l'atome d'oxygène du groupe carbonyle qui est une base dure, (h = 19 eV), est à l'origine du phénomène d'activation électrophile. La réaction est sous contrôle de charge.
- les organomagnésiens fournissent généralement un mélange de produits d'addition 1, 2 et 1, 4 ;
l'ajout de CuBr augmente considérablement l'addition 1, 4.
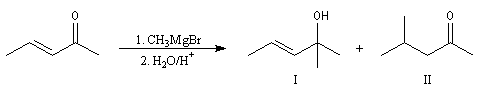 ComposésIIIsans CuBr9010avec CuBr595
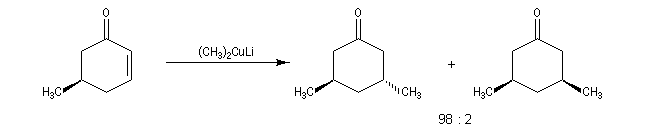
ComposésIIIsans CuBr9010avec CuBr595 - les cuprates lithiens de Gilman fournissent généralement de façon quasi exclusive le produit d'addition 1, 4.
La réaction s'effectue avec une grande stéréosélectivité.
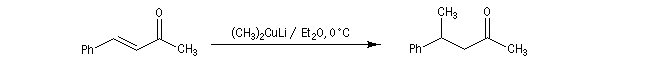
- Les réactifs de Knochel réagissent en position 4 sur les cétones a, b-insaturées en présence de chlorure de triméthylsilyle.
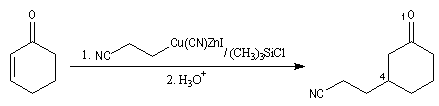
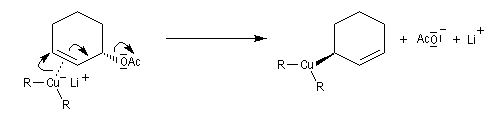
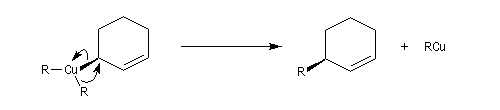
La molesse du carbone du carbone nucléophile du cuprate peut aussi être invoquée dans le cadre d'un contrôle orbitalaire de la réaction (Cu+ est un acide mou, de dureté de 6,9). L'attaque préférentielle de l'organométallique a lieu sur l'atome de carbone qui possède le plus gros coefficient dans l'orbitale BV, donc sur l'atome C4. Synthèse de la prostaglandine PGF2a
La synthèse stéréosélective de la prostaglandine PGF2a selon la méthode mise au point par G. Stork, constitue un exemple de l'utilisation des cuprates vinyliques en synthèse. Le cuprate s'additionne sur la face prochirale la moins encombrée de la double liaison éthylénique.
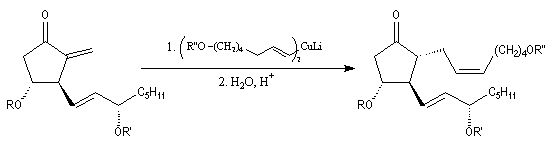
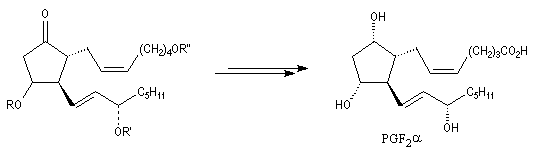
 |
La prostaglandine PGF2a est une molécule biologique possédant des effets physiologiques importants. Elle intervient dans le cycle ovarien et dans le mécanisme provoquant l'accouchement. Elle a également une activité antiinflammatoire. La synthèse totale de cette molécule a été réalisée par plusieurs équipes dont celle du chimiste américain E. J. Corey en 1970 ainsi que par celle de son compatriote d'origine belge, G. Stork selon une voie différente dont quelques étapes sont décrites plus haut. Le précurseur biochimique des prostaglandines est l'acide arachidonique. |
L'addition conjuguée d'un nucléophile sur un substrat a, b-éthylénique a été mise en évidence par le chimiste américain A. Michaël en 1887. Cette réaction, très précieuse pour réaliser des allongements de chaînes carbonées, met en jeu des nucléophiles variés : ion énolate, ion cyanure, dithiane lithié, énamine, ylures de soufre, carbanions issus de dérivés nitrés etc. De même des substrats divers peuvent être utilisés comme accepteurs de Michaël : aldéhydes, cétones, esters, nitriles, nitroalcènes etc. Leur point commun est de posséder une double liaison éthylénique rendue électrophile par la présence d'un groupe attracteur conjugué avec elle. Lorsque la réaction implique deux composés carbonylés, l'un jouant le rôle d'accepteur et l'autre de nucléophile, on obtient un système dicarbonylé 1,5. Dans le cas le plus simple cela donne le schéma de rétrosynthèse suivant :
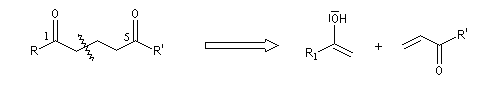
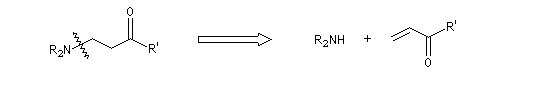
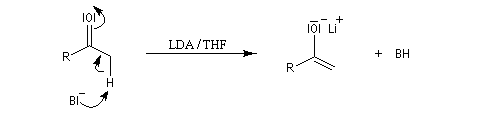
Examinons comme exemple le cas de l'addition d'un ion énolate sur un composé carbonylé. La préparation des énolates a été vue dans le chapitre relatif aux composés carbonylés.
- Une quantité catalytique de base suffit dans le cas où la formation de l'énolate
est réalisée sous contrôle thermodynamique.
- La réaction peut être réalisée sous contrôle cinétique si l'énolate est préformé par réaction entre le réactif de départ et un équivalent de base comme le LDA.

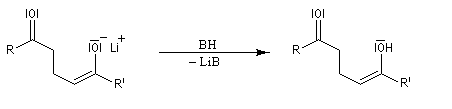
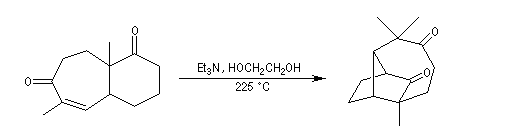
L'exemple suivant concerne une synthèse de la (R)-(-)-muscone, une cétone à grand cycle de structure voisine de celle de la civétone par Tanaka et al [42]. excès énantiomérique est de 99 %.
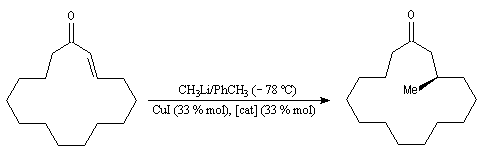
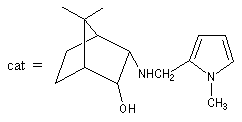
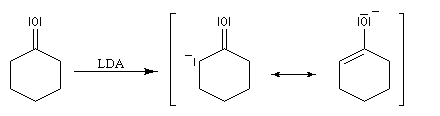
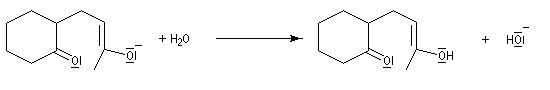
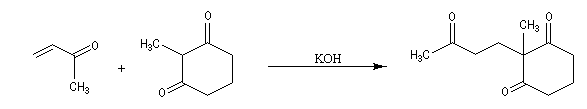
Annélation de Robinson L'annélation de Robinson (en anglais : Robinson annulation) est la combinaison d'une réaction de Michaël suivie d'une condensation aldol. Cette synthèse de cycles a été mise au point par le chimiste britannique R. Robinson en 1935. Voici un exemple de cette réaction qui utilise l'énolate de la cyclohexanone. On commence par préparer cet ion énolate en déprotonant la cyclohexanone par le LDA.
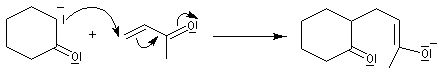
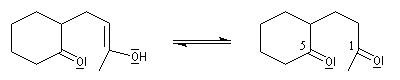
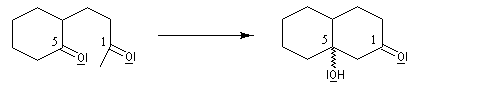
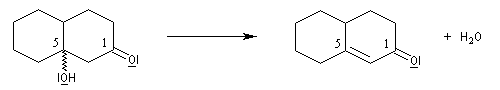
Application 1 : synthèse de la cétone de Wieland-Miescher
 |
La cétone de Wieland-Miescher sert de point de départ dans plusieurs synthèses de stéroïdes ou de terpènes comme celles du longifolène par E. J. Corey (1964), [39] et celle de J. E. Mc Murry et S. J. Isser (1972), [40]. |
Une synthèse énantiosélective utilisant la L-proline comme auxilliaire chiral a été mise au point. Il s'agit d'un cas particulier de la réaction de Hajos-Parrish au cours de laquelle le cétol n'est pas isolé du fait de la température de travail élevée. L'excès énantiomérique est voisin de 90 %.
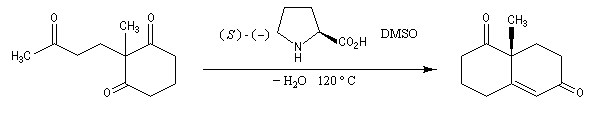
Le principe est le même que précédemment. Il y a cette fois quatre cycles à créer. On va donc utiliser deux fois successivement la suite de réactions : addition de Michaël suivie d'une cétolisation.
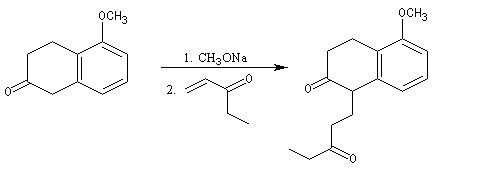
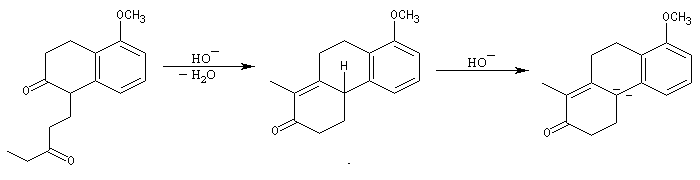
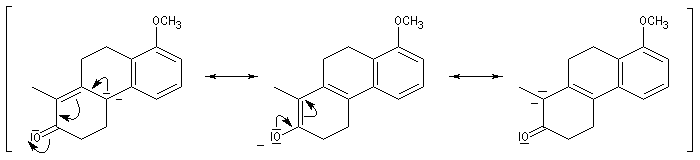
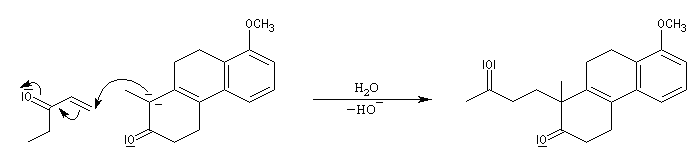
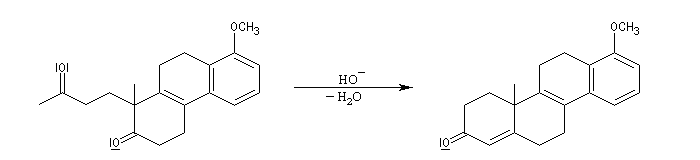
L'origine de cette réaction remonte aux travaux du chimiste japonais K. Morita en 1968. L'auteur utilisait une phosphine substituée par des groupes cyclohexyle comme catalyseur. Le rendement, très faible, (< 20 %) de la réaction limite les appplications possibles.
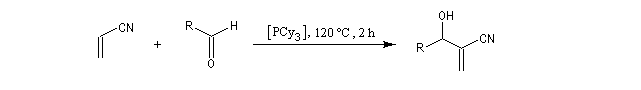
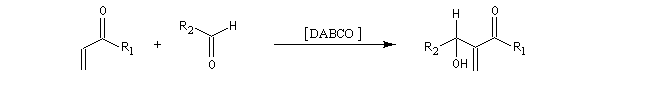
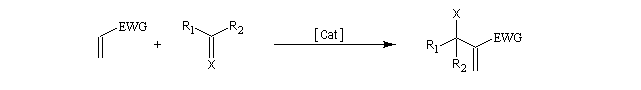
Il s'agit donc d'une méthode très générale de synthèse de composés acryliques.
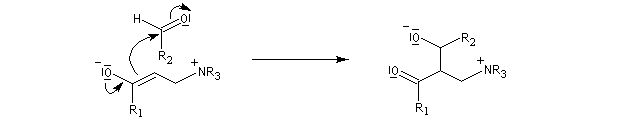
La première étape de la transformation est l'addition réversible d'une amine tertiaire comme DABCO, DBU ou encore la DMAP sur une a-énone, jouant le rôle d'accepteur de Michaël. Il y a formation d'un zwitterion.

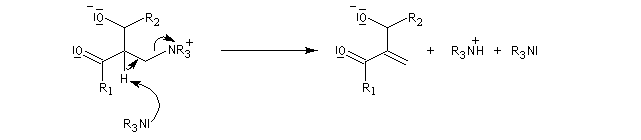

- en stabilisant le zwitterion ;
- en utilisant un aldéhyde plus électrophile.
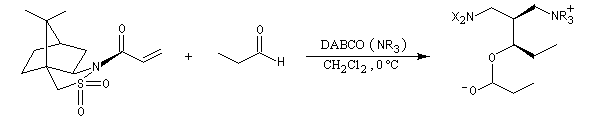
Un des grands intérêts de la réaction de Baylis Hillman provient du fait qu'il en existe des versions stéréosélectives.
La version diastéréosélective suivante utilise comme auxilliaire chiral un sultame d'Oppolzer.
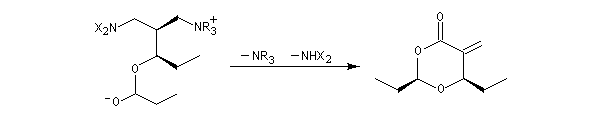
Bibliographie
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education.
[12] S. Hünig, E. Lücke, W. Brenninger, Organic Syntheses.
Ouvrages théoriques
[13] A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
[14] J. March - Advanced organic chemistry, Wiley Interscience.
[15] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
[16] V. Minkine - Théorie de la structure moléculaire, Editions Mir 1979.
[17] N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
[18] R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
[19] R. B. Woodward, R. Hoffmann, The conservation of Orbital Symmetry (Verlag Chemie, Weinheim 1970).
[20] P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
[21] P. Atkins - Molecular Quantum Mechanics, Oxford University Press 1983.
[22] J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.
[23] P. -G. de Gennes et J. Badoz - Les objets fragiles, Plon 1994.
[38] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley 1976.
Liens
[24] Organic Pericyclic reaction by H. Rzepa at the Department of Chemistry Imperial College
[25]
[26] Eléments de chimie quantique
[27] The synthesis and reactivity of Cubane by Beinn Muir
[28] Diels-Alder reaction
[29]
[30]
[31] O. Diels and K. Alder the Nobel Prize in Chemistry 1950
[32] K. Fukui and R. Hoffmann the Nobel Prize in Chemistry 1981
[33]
[36] Michaël addition
[37] Robinson annulation
[41] Diels-Alder reaction an animation
[43] K. Ziegler and G. Natta the Nobel Prize in Chemistry 1963
[44]
[45] Wieland Miescher Ketone by Paul Buchschacher, A. Fürst, and J. Gutzwiller
[46] The Diels-Alder Reaction in Total Synthesis by K. C. Nicolaou.
[50] The development of Intrinsically Conducting Polymers by Mats Anderson
[51] The Nobel Prize in Chemistry 2000
[52] Thèse F. Arnaud
[53] The Morita-Baylis-Hillman reaction by Stephen Goble
[54] Quelques applications de la méthode des orbitales frontières
[56] Introduction à la chimie moléculaire par les orbitales frontières Pascal le Floch, Ecole Poytechnique.
[57] Steroïd Industry Honored
[58] Molecule of the month : Taxol
[59] The conservation of Orbital Symmetry by Rob Knowles MacMillan Group Meeting February 2nd, 2007
Articles
[34] Aromaticity, Special Publication N° 21, The Chemical Society, London (1967).
[35] R. B. Woodward, R. Hoffmann ; J. Am. Chem. Soc., 87, 395 (1965).
[39] Corey, E. J. ; Mitra, R. B. ; Vatakencherry, P. A. ; J. Am. Chem. Soc. 86, 478 (1964).
[40] McMurry, Isser S. J. ; J. Am. Chem. Soc., 94, 7132 (1972).
[42] K. Tanaka, J. Matsui, H. Suzuki, J. Chem. Soc., Perkin Trans. 1, 1993, 15.
[47] J. Rakowitz, Excitant oxygène singulet, Bulletin de l'Union des Physiciens, novembre 1984.
[48] G. Levy, L'oxygène ou le rouge émis, Bulletin de l'Union des Physiciens, mai 1982.
[55] Steven H. Bertz, Anu Chopra, Magnus Eriksson, Craig A. Ogle, and Paul Seagle. J. Am. Chem. Soc., 124 (46), 13650 - 13651, 2002. 10.1021.
[60] W. S. Rapson, R. Robinson, J. Chem. Soc. 1935, 1285
[61] Wieland, P.; Miescher, K. Helv. Chim. Acta 1950, 33, 2215–2228.
[62] Nicolaou, KC ; Yang, Z ; Liu, JJ ; Ueno, H; Nantermet, PG ; Guy, RK ; Claiborne, CF; Renaud, J et al. (February 1994) Nature 367 (6464): 630–634.









0 commentaires:
Enregistrer un commentaire