Cours de chimie organique -
Alcènes et autres composés éthyléniques
Généralités
Nomenclature
On appelle alcènes, les hydrocarbures insaturés non cycliques de formule CnH2n. La chaîne principale est celle qui comporte le plus grand nombre de liaisons et, le cas échéant, le plus grand nombre d'atomes de carbone. Le nom du composé dérive de celui de l'alcane en remplaçant la terminaison ane par ène. La position de la double liaison est indiquée par le numéro de l'atome de carbone doublement lié qui possède l'indice le plus petit. La numérotation est effectuée de façon que la double liaison ait l'indice le plus faible.
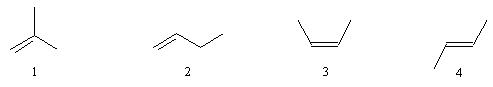
|
1
|
2
|
3
|
4
|
|
méthylpropène
|
but-1-ène
|
(Z)-but-2-ène
|
(E)-but-2-ène
|
Quelques alcènes importants
L'éthène est l'alcène le plus important sur le plan
industriel. Il est produit par craquage d'hydrocarbures. Environ la
moitié de l'éthène
produite est utilisée pour préparer les polyéthylènes (basse densité et
haute densité). L'autre moitié permet la
préparation de composés importants :
|
Le propène est le deuxième composé éthylénique le plus
important comme produit de base de l'industrie chimique. Il est produit
par craquage d'hydrocarbures. La part la
plus importante sert à la synthèse du polypropène (polypropylène). Le
reste permet la préparation des composés suivants :
|
Les terpènes constituent un groupe important de composés éthyléniques naturels. Ils dérivent formellement de l'isoprène
|
La molécule de gauche est celle de longifolène, un
sesquiterpène naturel produit par plusieurs variétés de conifères. C'est
en effectuant la synthèse de cette molécule que le
chimiste américain E. J. Corey (prix Nobel de Chimie 1990) développa
pour la première fois la notion d'analyse rétrosynthétique (1957) [64].
Display : spacefill ball & stick sticks wireframe Zoom : in out |
Les cyclènes sont des hydrocarbures de formule CnH2n-2. Les cycles moyens possédant 5 ou 6 atomes de carbone possèdent une réactivité comparable à celle des alcènes. Certains cyclènes possèdent un plan de chiralité. Le trans-cyclooctène est dédoublable en deux énantiomères.
Diènes, polyènes
Les diènes sont des composés éthyléniques qui possèdent deux liaisons doubles. Leur étude est faite dans le chapitre consacré aux composés conjugués. A côté des diènes dont les doubles liaisons sont éloignées et qui se comportent comme des éthyléniques ordinaires, on distingue :
- les diènes conjugués qui possèdent une réactivité originale ;
- les diènes cummulés, encore appelés allènes. Les allènes dissymétriques présentent le phénomène de chiralité axiale.
Le barrelène (bicyclo[2.2.2]octa-2,5,7-triène) est un
composé éthylénique synthétisé par le chimiste américain H. E. Zimmerman
en 1960.
Display : spacefill ball & stick sticks wireframe Zoom : in out |
Structure de l'éthène
La simple connaissance de la formule brute C2H4 est en faveur d'une liaison double entre les atomes de carbone. Le tableau suivant rassemble des données expérimentales relatives à l'éthane et à l'éthène.
|
Liaison
|
Longueur (nm)
|
Energie (kJ.mol-1)
|
|
C-C
|
0,154
|
347
|
|
C=C
|
0,133
|
606
|
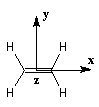 |
Les expériences de diffraction montrent que les atomes de carbone et d'hydrogène sont situés dans un même plan. La molécule
présente trois plans de symétrie :
|
Les atomes impliqués dans la liaison double ont une géométrie localement plane. L'existence de la double liaison chez les alcènes bloque la libre rotation des groupes qui lui sont attachés. Elle est à l'origine de la diastéréo-isomérie Z, E.
Notons que l'isomérisation photochimique du rétinal de stéréochimie E en néorétinal de stéréochimie Z est à la base du mécanisme de la vision.
Constantes physiques
Les alcènes ont des propriétés physiques assez comparables à celles des alcanes. Il est intéressant de noter des différence de propriétés assez fines entre des alcènes stéréoisomères comme l'illustre le cas des but-2-ène. Ces différences de propriétés physicochimiques permettent de distinguer expérimentalement ces stéréoisomères.
|
composé
|
moment dipolaire (D)
|
température d'ébullition (°C)
|
|
(Z)-But-2-ène
|
0,4
|
4
|
|
(E)-But-2-ène
|
0
|
1
|
Niveaux d'énergie, formalisme simplifié s, p
Les considérations de symétrie précédentes sont très importantes pour l'application de la théorie des orbitales moléculaires. Une simplification importante intervient pour les molécules qui comme l'éthène sont planes. La réflexion par rapport au plan de la molécule (xOy) permet de séparer les orbitales moléculaires en deux groupes :
- celles qui sont inchangées dans la réflexion par rapport au plan de la molécule, qui sont appelées orbitales s ;
- celles qui changent de signe dans la réflexion par rapport à ce plan, qui sont appelées orbitales p.
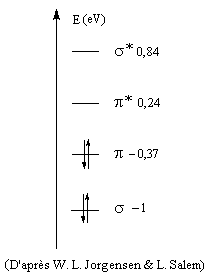 |
L'étagement des niveaux d'énergie moléculaires peut être obtenu par le calcul. Il est possible de le vérifier expérimentalement dans une certaine mesure en utilisant les techniques de spectroscopie photoélectronique et de spectroscopie électronique pour l'analyse chimique (ESCA). Le diagramme complet des orbitales moléculaires de l'éthène comporte 12 niveaux. L'image de gauche représente un diagramme simplifié ne comportant que les orbitales de symétrie p et celles de symétrie s respectivement liante et antiliante pour tous les atomes de la molécule.
|
L'étude de la molécule d'éthène a été faite dans le chapitre consacré à la méthode de Hückel simple (HMO) et en utilisant la méthode de Hückel étendue (EHMO)
Le tableau suivant regroupe les valeurs numériques des énergies et des coefficients des orbitales atomiques dans les orbitales moléculaires (HMO) :
|
Energie
|
Ci1
|
Ci2
|
|
E1 = a + b
|
0,707
|
0,707
|
|
E2 = a - b
|
0,707
|
-0,707
|
Les orbitales moléculaires (HMO) ont pour expression :
La plus haute orbitale moléculaire occupée et la plus basse orbitale vacante sont appelées orbitales frontières. La connaissance des énergies et des propriétés de symétrie de ces orbitales moléculaires est particulièrement intéressante pour étudier la réactivité de la molécule dans le cas où la réaction est sous contrôle orbitalaire. Les électrons p sont les plus mobiles et les plus facilement polarisables lors de l'interaction de la molécule avec un réactif extérieur.
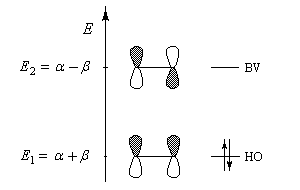 |
Les orbitales frontières de la molécule d'éthène :
|
Spectroscopie
Spectroscopie infrarouge
La vibration de valence de la double liaison absorbe vers 1650 cm-1. Les fréquences d'absorption des vibrations de déformation des liaisons C-H sont également précieuses pour identifier les structures. Quelques absorptions caractéristiques sont données ci-dessous.
|
Composé
|
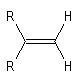 |
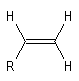 |
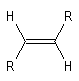 |
|
s (cm-1)
|
890
|
915 & 995
|
970
|
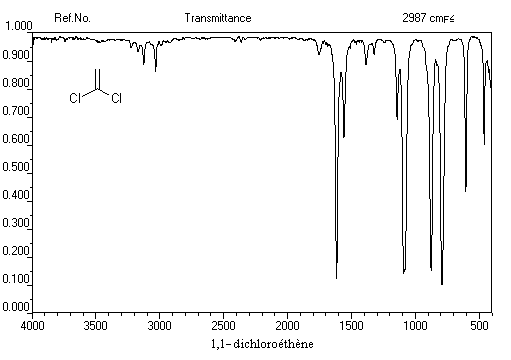
- Les quatre protons de l'éthène donnent un pic unique pour un déplacement chimique : d = 5,6 ppm. D'une façon générale, les atomes d'hydrogène liés à la double liaison appelés hydrogènes vinyliques, ont des déplacements chimiques assez élevés.
Cette propriété est liée à la circulation des électrons p dans le champ magnétique extérieur qui induit un champ opposé au champ appliqué dans la région où se trouvent ces protons.
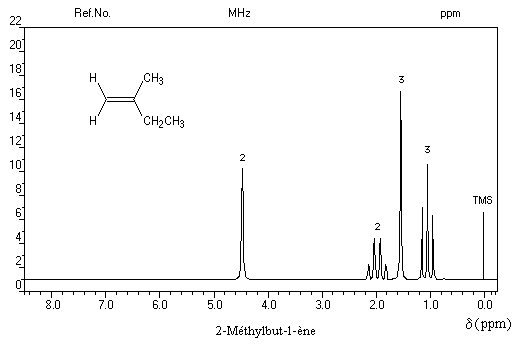
- La spectroscopie de RMN permet de distinguer facilement les diastéréo-isomères possédant des atomes d'hydrogène
vicinaux grâce à la valeur plus élevée de la constante de couplage 3J pour l'isomère E par rapport à l'isomère Z. Les grandeurs numériques suivantes n'ont qu'une valeur indicative.
 constante de couplage2J = 13J = 103J = 16
constante de couplage2J = 13J = 103J = 16
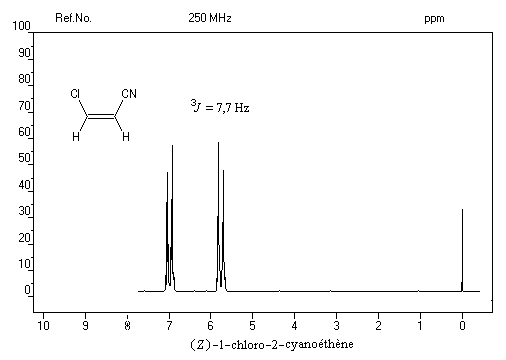 Dans le spectre précédent, on notera que les doublets ne sont pas symétriques (effet de toit). Cela vient du fait que le couplage entre les protons est un couplage fort.
Dans le spectre précédent, on notera que les doublets ne sont pas symétriques (effet de toit). Cela vient du fait que le couplage entre les protons est un couplage fort.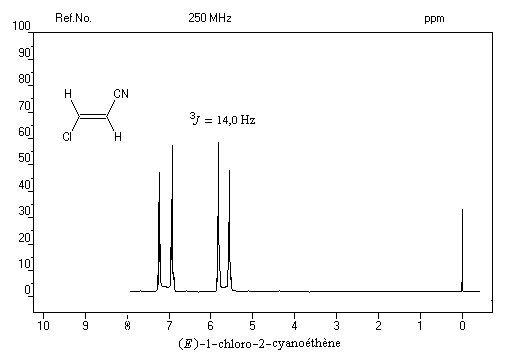
Un autre exemple est celui de la molécule d'acide (E) cinnamique. - Les spectres des alcènes monosubstitués sont complexes lorsque le déplacement chimique des protons et la constante de couplage sont du même ordre de grandeur.
ComposéJABJACJBCJ (Hz)10152Les signaux des protons A, B et C ont l'allure suivante.
Fréquemment, l'analyse au premier ordre n'est possible qu'avec des spectromètres fonctionnant à haute fréquence (600 MHz).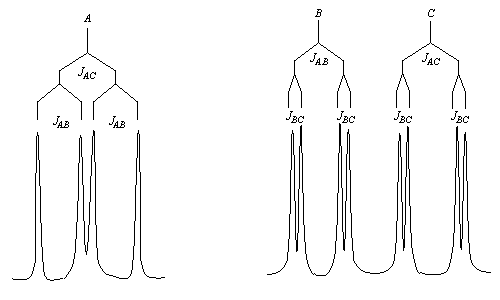
Conditions expérimentales
La réaction entre le dihydrogène H2 et un composé éthylénique est une réaction d'addition. Elle est interdite pour des raisons de symétrie orbitalaire et n'est réalisable à une vitesse raisonnable qu'avec l'aide d'un catalyseur hétérogène ou un catalyseur homogène permettant de lever cette interdiction. D'un point de vue thermodynamique, elle est quasi-totale dans les conditions normales de température et de pression. Le rendement est de l'ordre de 90 %.
Puisqu'un catalyseur accélère aussi bien la réaction directe que la réaction inverse, la déshydrogénation d'un hydrocarbure saturé est également envisageable mais elle constitue surtout une méthode industrielle en raison de son manque de sélectivité.
Comparaison de la stabilité des alcènes
La réaction d'hydrogénation peut être mise à profit pour évaluer la différence de stabilité entre alcènes stéréoisomères. Raisonnons sur les diastéréoisomères Z et E du but-2-ène.
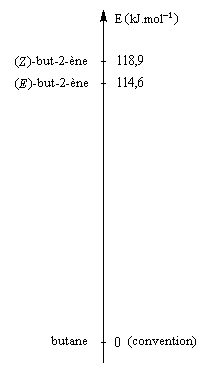 |
L'hydrogénation du (Z)-but-2-ène dégage une énergie de -118,9 kJ.mol-1. Celle du (E)-but-2-ène dégage -114,6 kJ.mol-1. On en déduit l'enthalpie de la réaction suivante : 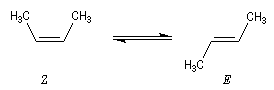
-118,9 - (-114,6) = - 4,3 kJ.mol-1
Le stéréoisomère E est donc plus stable que le stéréoisomère Z |
 |
 |
L'addition de H2 sur le 3, 4-diméthylhex-3-ène de configuration Z fournit de façon quasi-exclusive le (3R, 4S)-3, 4-diméthylhexane achiral.
Le (3R, 4R)-3, 4-diméthylhexane et son énantiomère le (3S, 4S)-3, 4-diméthylhexane, tous trois diastéréo-isomères du précédent ne sont pas obtenus. La réaction est donc diastéréosélective. Les atomes d'hydrogène se sont fixés du même côté de la double liaison. La stéréochimie de l'addition est syn.
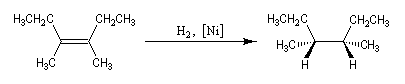
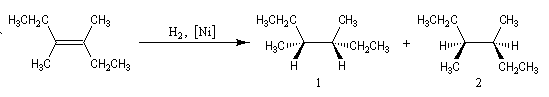
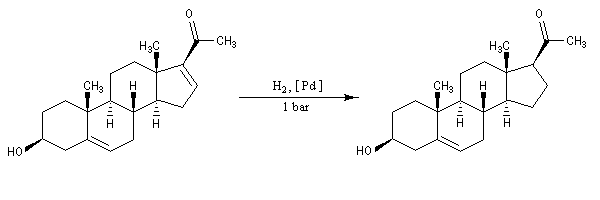
Dans l'exemple ci-dessous, impliquant un composé bicyclique ponté, l'hydrogène se fixe préférentiellement sur la face exo la moins encombrée.
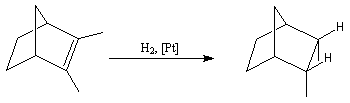
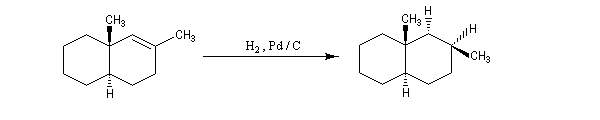
L'hydrogénation catalytique utilisant un métal de transition a été découverte par le chimiste français P. Sabatier (prix Nobel 1912 conjointement avec G. Grignard) [39]. Sabatier utilisait du nickel préparé par réduction de NiO par le dihydrogène.
Le nickel de Sabatier est surtout utile quand on veut effectuer des hydrogénations sélectives. Des catalyseurs plus actifs sont utilisés :
- le platine ou le palladium déposés sur du carbone ;
- le nickel de Raney est une autre variété de nickel activé (voir plus bas);
- le catalyseur d'Adams est du dioxyde de platine PtO2, réduit en Pt colloïdal par le dihydrogène in situ. Voici un exemple d'application intervenant dans la synthèse de R von Schleyer de l'adamentane.
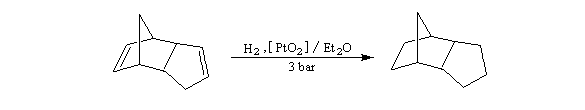
 |
Le Nickel de Raney, mis au point par l'ingénieur américain Murray Raney, est préparé en attaquant un alliage Ni-Al par une solution aqueuse d'hydroxyde de sodium. L'aluminium, fortement réducteur, réduit l'eau en milieu basique et passe en solution pour donner des ions complexes [Al(OH)4]-. Il reste le nickel sous forme d'une poudre noire très fine, possédant une grande surface active. Les termes anglais : spongy nickel ou skeletal nickel désignant ce catalyseur, reflètent bien sa structure alvéolaire. Le nickel de Raney doit être conservé dans l'eau car il est pyrophorique. (photographie G. D.) |
Le caractère syn de l'addition peut être interprété grâce au modèle suivant dans lequel les réactifs adsorbés à la surface du catalyseur sont dans une disposition géométrique favorable pour la réaction d'addition.
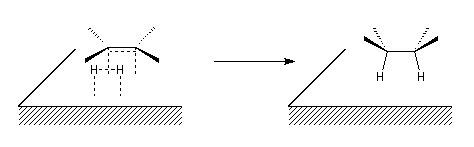
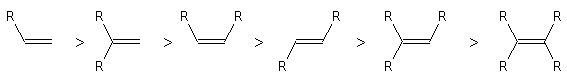

L'hydrogénation peut aussi être réalisée en milieu homogène en utilisant un catalyseur soluble comme le catalyseur de Wilkinson. La réaction d'addition est stéréospécifique, de stéréospécificité syn.
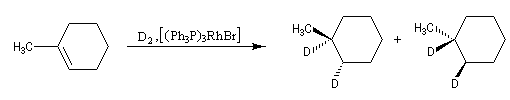
 |
L'image de gauche représente un modèle compact de [RhCl(PPh3)3]. Ce composé de couleur rouge-violet a été
synthétisé pour la première fois par G. Wilkinson et J. A. Osborn en 1966. Il peut être préparé par réaction entre la triphénylphosphine et le trichlorure de rhodium dans l'éthanol.
|
L'hydrogénation d'un composé éthylénique comportant des atomes de carbone prochiraux peut être effectuée de façon énantiosélective en présence de catalyseurs chiraux solubles. De tels catalyseurs sont capables de distinguer les faces énantiotopiques d'une double liaison. Le premier exemple de réaction de ce type est dû au chimiste américain W. S. Knowles en 1968. Il concerne l'hydrogénation d'un dérivé substitué du styrène.
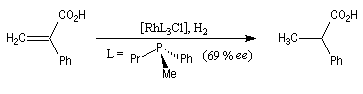
Plusieurs catalyseurs d'hydrogénation énantiosélective ont été mis au point ces dernières années. Citons deux exemples :
- le catalyseur utilisé dans la synthèse de la L-DOPA décrite ci-dessous, est un complexe du rhodium dérivé du catalyseur de Wilkinson, possédant une diphosphine chirale comme ligand ;
- des complexes du ruthénium sont également utilisés. Le complexe [Ru(R-BINAP)(tBuCOO)2] introduit par le chimiste japonais R. Noyori, un des grands spécialistes mondiaux de la synthèse énantiosélective, comporte un ligand binaphtyle chiral. Ce ligand peut exister sous deux formes énantiomères atropisomères. Un exemple d'hydrogénation énantiosélective d'un alcool allylique, le (E)-3, 7-diméthyl-2, 6-octadièn-1-ol ou géraniol, est donné à la référence [23].
La L-DOPA ou acide acide 2-amino-3-(3,4- dihydroxyphényl)-propanoïque est le précurseur d'un neuromédiateur, la dopamine, dont la déficience est à l'origine de la maladie de Parkinson. C'est l'un des seuls médicaments dont on dispose actuellement pour lutter contre cette maladie. Sa synthèse a été réalisée par W. S. Knowles en 1975 (prix Nobel de chimie 2001 avec K. B. Sharpless et R. Noyori ; [20]). Le procédé mis au point par Knowles pour Monsanto a permis une préparation à l'échelle industrielle à partir de la vanilline. Ce procédé n'est plus utilisé de nos jours.
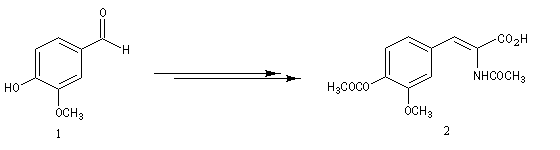
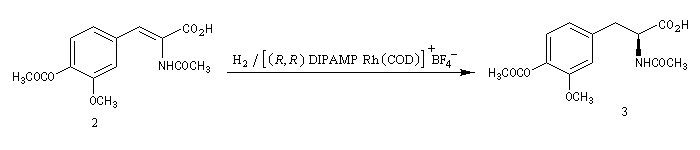
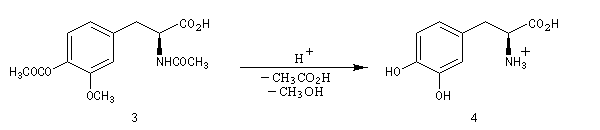
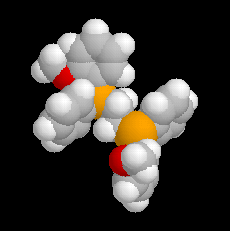 |
L'image de gauche représente la diphospine DIPAMP, ligand chiral d'un complexe du ruthénium utilisé par W. S. Knowles dans la synthèse industrielle de la L-DOPA représentée ci-dessous : 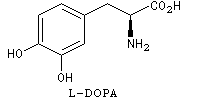 |
Résultats expérimentaux
L'une des premières synthèses industrielles du dichloroéthane, qui est un intermédiaire dans la synthèse du chlorure de vinyle, consistait en une réaction d'addition entre l'éthène et le dichlore.
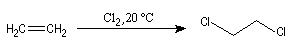
Les composés éthyléniques additionnent facilement Cl2 et Br2 pour conduire à des dérivés dihalogénés vicinaux. F2 très oxydant détruit la molécule. Avec I2 peu réactif, la réaction conduit à un équilibre défavorable au produit. La réaction s'effectue par union directe dans les conditions normales de température et de pression. Les solvants utilisés sont CH2Cl2, CHCl3, CCl4, AcOH.
Test analytique des doubles liaisons éthyléniques
La réaction de bromation peut être utilisée comme test des doubles liaisons éthyléniques. Sur la photographie (I) l'erlenmeyer contient une solution de dibrome Br2 dans CCl4 (on observe les vapeurs de Br2 qui s'échappent de la solution). L'ajout de quelques gouttes d'hex-1-ène provoque une décoloration rapide de la solution (photos II et III).
 |
 |
 |
|
I
|
II
|
III
|
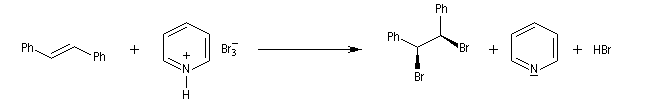
Le tribromure de pyridinium présente une alternative intéressante à l'utilisation du dibrome en raison de sa simplicité d'emploi.
Stéréochimie
L'addition de Br2 sur le (E)-but-2-ène fournit quasi exclusivement le (2R, 3S)-dibromo-2,3-butane. Ce composé est achiral car la molécule possède un plan de symétrie. Il s'agit d'un composé méso. Puisqu'on obtient un seul stéréoisomère parmi plusieurs pouvant se former a priori, la réaction de bromation est diastéréosélective.
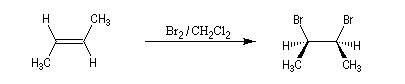
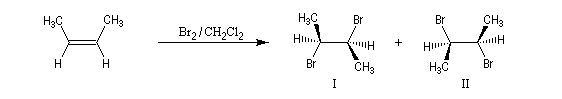
Mécanisme
Il s'agit d'un mécanisme par stades :
- L'étape cinétiquement déterminante implique l'attaque de la molécule de dibrome polarisable par les électrons p
de la
double liaison éthylénique. Elle conduit à la formation d'un ion
bromonium ponté (dans l'exemple choisi, cet ion est achiral ; il s'agit
d'un composé méso.)
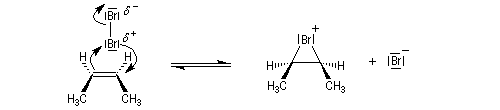
- L'ouverture de l'ion bromonium par l'ion bromure nucléophile se
produit dans une étape ultérieure. Elle ne peut avoir lieu pour des
raisons stériques que
du côté opposé au pont. Les réactions sur les atomes de carbone 1 et 2
sont équiprobables. On obtient le mélange mélange racémique des dibromures énantiomères I et II.
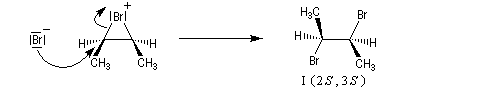
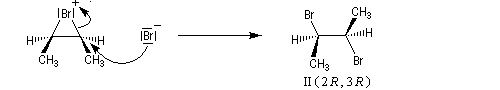
Ce mécanisme rend compte de la stéréospécificité anti de la réaction. Ions halonium
Les ions bromonium sont des intermédiaires très réactifs qui ne sont généralement mis en évidence que par des méthodes spectrales. Certains ont pu être isolés comme celui de l'exemple ci-dessous.
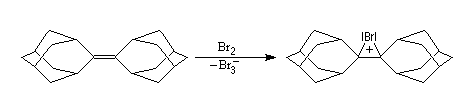
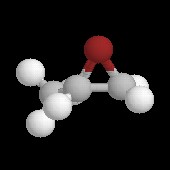 |
L'intervention d'ions bromonium dans les réactions de
bromation a pu, dans certains cas, être démontrée de façon directe. G.
Olah (1968) a ainsi mis en évidence par spectroscopie de RMN, les ions bromonium formés à partir du 1-bromo-2-fluoropropane en milieu superacide [61].
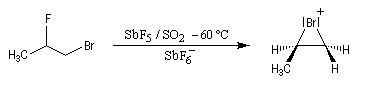 |
- Dans l'exemple suivant, la bromation du (Z)-1-phénylpropène, fournit majoritairement, mais non exclusivement, le mélange racémique des énantiomères du couple like.
On envisage la formation d'ions bromonium intermédiaires.
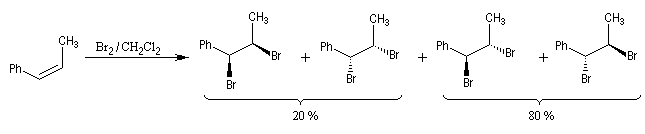 La baisse de stéréosélectivité s'explique par l'intervention d'un carbocation benzylique stabilisé par résonance. L'ion bromure peut alors réagir au dessus ou au dessus du cation localement plan.
La baisse de stéréosélectivité s'explique par l'intervention d'un carbocation benzylique stabilisé par résonance. L'ion bromure peut alors réagir au dessus ou au dessus du cation localement plan.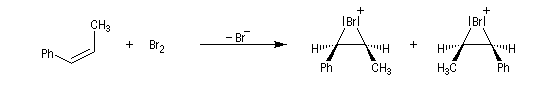
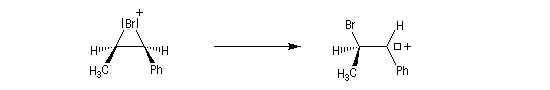
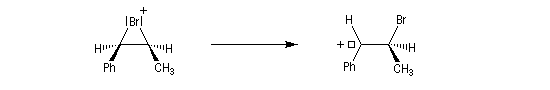
- Dans le cas de la chloration, la formation de l'ion ponté est défavorisée par rapport à celle du carbocation. On attribue cette différence à la taille plus petite de l'atome de chlore comparée à celle de l'atome de brome. La formation du pont est beaucoup plus difficile.
- La formation d'ions iodonium est possible mais la réaction d'iodation des liaisons éthyléniques et thermodynamiquement défavorisée.
Dans l'iodolactonisation, un acide carboxylique g-insaturé est cyclisé en lactone substituée.
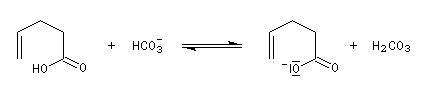
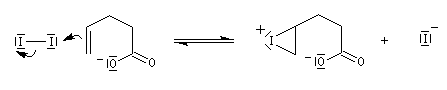
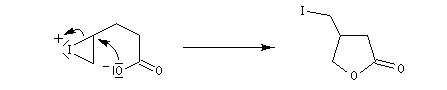
Protection des doubles liaisons éthyléniques
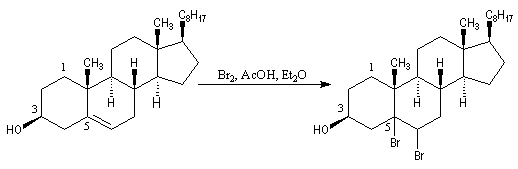
L'oxydation du cholestérol en milieu acide, pose le problème de l'isomérisation de la cholest-5-ène-3-one en cholest-4-ène-3-one, une cétone conjuguée plus stable [16] . Une solution consiste à bloquer la double liaison éthylénique.
- l'addition du dibrome conduit au dérivé dibromé ;
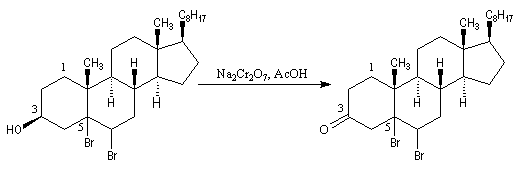
- l'oxydation de la fonction alcool secondaire conduit à la formation d'une fonction cétone ;
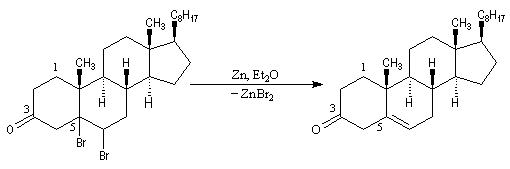
- la double liaison éthylénique est régénérée grâce à une réaction d'élimination, entre le composé précédent et le zinc.
Utilisation du tribromure de pyridinium
Le tribromure de pyridinium est un agent bromant introduit par C. Djerassi en 1948 [56]. Dans la littérature anglo-saxone, le tribromure de pyridinium est généralement appelé : "pyridinium bromide perbromide" (en abrégé PBP). Les ions tribromure ont une structure analogue à celle des ions triodure, c'est à dire quasi linéaire. Dans la méthode VSEPR, ils correspondent au type AX2E3.
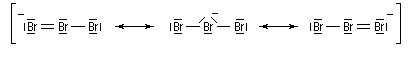
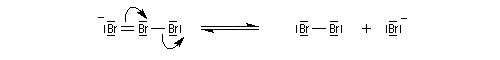
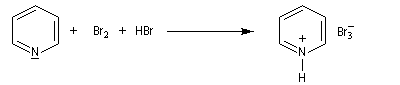
Le chlrorure d'iode ICl (réactif de Wijs) s'additionne rapidement et de façon quantitative sur les doubles liaisons éthyléniques.
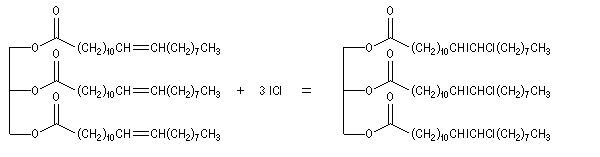
|
|
Le réactif de Wijs peut être préparé en faisant réagir le trichlorure d'iode ICl3 (7,8 g) et le diiode I2
(8,5 g) dans 250 mL d'acide acétique pur (glacial) à chaud. Après
refroidissement, on complète le mélange à 1 L avec de l'acide acétique
pur.
|
Résultats expérimentaux
La réaction entre HBr dissous dans l'acide éthanoïque et le méthylpropène à 20 °C donne essentiellement du 2-bromo-2-méthypropane (1) et seulement des traces de 1-bromo-2-méthylpropane (2).
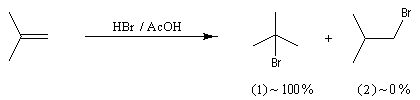
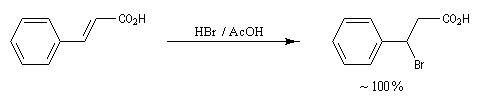
Il s'agit d'un mécanisme par stades.
- Une petite quantité d'hydracide est dissociée dans le solvant :
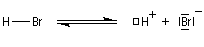
- La réaction électrophile de la double liaison sur le proton conduit à la formation d'un carbocation. C'est l'étape cinétiquement déterminante de la réaction.
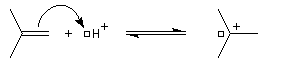
- La réaction entre le carbocation et l'ion bromure nucléophile s'effectue dans une seconde étape. Elle peut avoir lieu indifféremment sur l'une ou l'autre
face de l'atome de carbone trigonal. La réaction n'est donc pas stéréosélective.
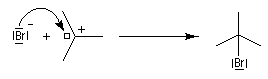

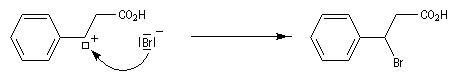
Puisque le mécanisme fait intervenir un carbocation, on peut observer des réactions de transposition de type Wagner-Meerwein. C'est le cas, par exemple, dans la réaction suivante de préparation de l'acétate d'isobornyle à partir du camphène qui constitue une étape de la synthèse industrielle du camphre.

Résultats expérimentaux
L'addition d'eau sur un composé éthylénique s'appelle hydratation. Elle conduit à la formation d'un alcool. Avec les alcènes substitués qui forment facilement des carbocations, il suffit d'utiliser un acide dilué. L'ion H+ est alors un catalyseur de la réaction :
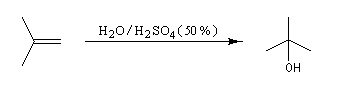
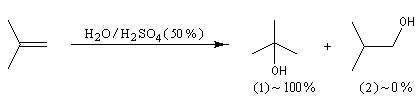
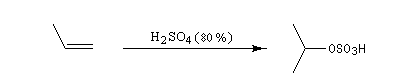
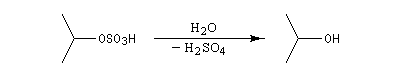
Il ressemble beaucoup à celui décrit pour l'addition de HBr. Il s'agit d'un mécanisme par stades. On peut regarder cette réaction comme la réaction inverse de la déshydratation des alcools.
- Raisonnons dans le cas du méthylpropène. La première étape
est l'attaque électrophile de la double liaison sur un proton servant de
catalyseur.
Elle conduit à la formation d'un carbocation. C'est l'étape cinétiquement déterminante.
- La réaction entre le carbocation et l'eau a lieu dans une deuxième étape rapide.
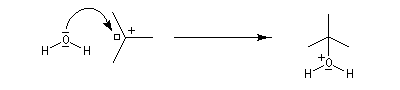
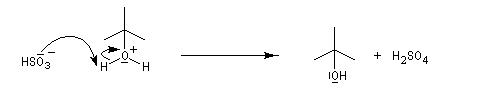
L'hydratation telle que nous venons de la décrire n'est pas un procédé utilisé en synthèse. On préfère utiliser l'oxymercuration qui est beaucoup plus régiosélective et non sujette aux réactions de transpositions. Hydratation énantiosélective de l'acide fumarique
L'hydratation de l'acide (E)-butanedioïque ou acide fumarique constitue l'une des étapes du cycle de Krebs. La réaction est énantiosélective. Le produit de cette réaction est l'acide (2S)-2-hydroxybutanedioïque ou (L-malique). Afin de préciser le mécanisme de la réaction, l'hydratation a été effectuée en utilisant de l'eau lourde.
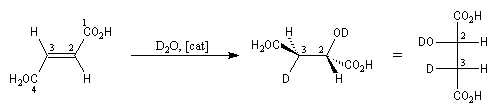
Enoncé historique
Cette règle énoncée par le chimiste russe V. V. Markovnikov en 1868 constitue l'une des premières et des plus célèbres tentatives de rationalisation de résultats expérimentaux en chimie organique. Il s'agissait à l'origine d'une règle empirique qui concernait l'addition des hydracides halogénés sur les alcènes dissymétriques. Markovnikov avait reconnu le caractère très régiosélectif de cette addition. La règle permet de prévoir l'atome de carbone sur lequel se fixe l'hydrogène de l'hydracide :
Lors de l'addition d'un hydracide sur un alcène dissymétrique l'atome d'hydrogène se fixe sur l'atome de carbone le moins substitué.
Examinons l'exemple de l'addition de HBr sur le méthylpropène. On obtient effectivement de façon quasi exclusive le 2-méthyl-2-bromopropane.
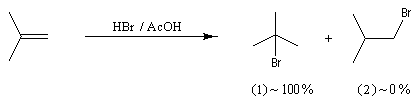
Le problème se pose de savoir si la régiosélectivité observée est le fruit d'un contrôle cinétique ou d'un contrôle thermodynamique de la réaction. Il est possible d'étudier l'équilibre entre les dérivés halogénés P1 et P2 et la constante de cet équilibre a été déterminée expérimentalement. On trouve : K = 4,5. Si la réaction était sous contrôle thermodynamique on obtiendrait une quantité non négligeable du produit P2 compte-tenu de cette valeur de K. Plus précisément, on aurait les pourcentages suivants :
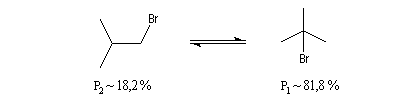
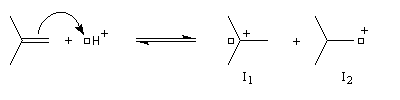
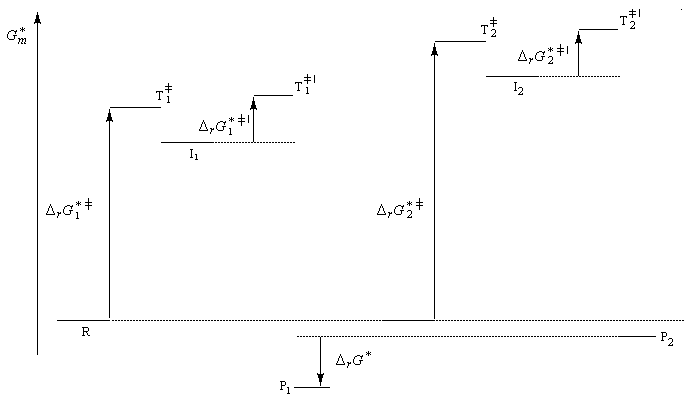
L'expérience montre que de nombreuses additions de réactifs dissymétriques s'effectuent sous contrôle cinétique par un mécanisme ionique impliquant un carbocation ou un ion positif ponté. En plus de l'addition des hydracides et de l'hydratation déjà cités citons l'addition de dérivés interhalogénés comme ICl ou des acides hypohalogéneux (ClOH, BrOH). Schématisons par Ad -- Hd+ le réactif dissymétrique. La règle de Markovnikov peut être généralisée :
Lors de l'addition d'un réactif Ad -- Hd+ sur un composé éthylénique dissymétrique, Ad - se fixe préférentiellement sur l'atome de carbone qui stabilise le mieux une charge positive.
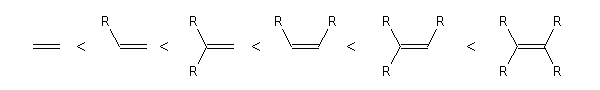
Lorsque le composé éthylénique est un alcène, l'atome de carbone qui stabilise le mieux une charge positive est celui qui est substitué par le plus grand nombre de groupes alkyles. Dans le cas des alcène substitués, la méthode des perturbations fournit un cadre théorique commode pour rationaliser ce résultat. Pour les alcènes fonctionnalisés, la situation est plus délicate et il faut examiner chaque cas.
Interprétation électronique de la règle de Markovnikov
Nous allons raisonner dans le cas du propène en appliquant les résultats de la méthode des perturbations. Le propène peut être considéré comme résultant de l'interaction entre une molécule d'éthène et un fragment méthyle. Ce dernier sera considéré dans le modèle à deux électrons et décrit par une orbitale d'énergie a + 2 b. Pour l'éthène, nous prendrons les orbitales moléculaires obtenues à partir de la méthode de Hückel simple.
Le calcul complet par la méthode des perturbations est effectué aux références [72] et [74]. Le diagramme d'énergie est le suivant.
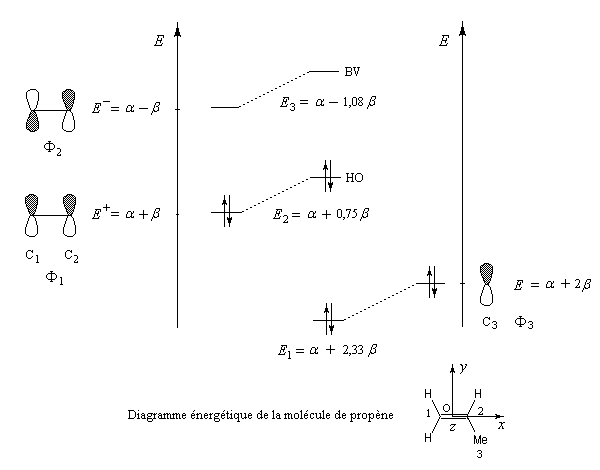
|
Energie
|
Ci1
|
Ci2
|
Ci3
|
|
E3 = a - 1,08b
|
- 0,668
|
0,726
|
- 0,165
|
|
E2 = a + 0,75b
|
0,707
|
0,552
|
- 0,441
|
|
E1 = a + 2,33b
|
0,207
|
0,413
|
0,877
|
- le niveau énergétique de la HO du propène est supérieur à celui de l'éthène. Le propène va être plus réactif vis à vis d'un électrophile que l'éthène ;
- en ce qui concerne la régiosélectivité un électrophile réagit de façon préférentielle sur l'atome de carbone dont le coefficient est le plus élevé dans la HO. Comme le montre le tableau précédent, il s'agit de l'atome de carbone C1 (coefficient : 0,707). C'est bien la règle de Markovnikov.
Résultats expérimentaux
La réaction entre l'acide hypobromeux et le 2-méthylbut-2-ène fournit un mélange mélange racémique de (3S)-3-bromo-2-méthylbutan-2-ol et (3R)-3-bromo-2-méthylbutan-2-ol énantiomères.
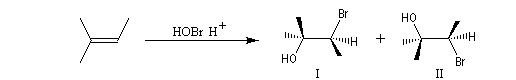
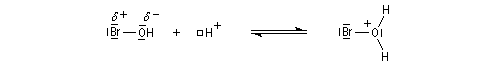
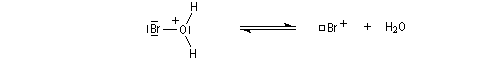
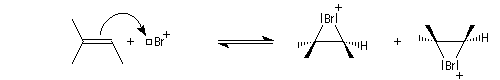
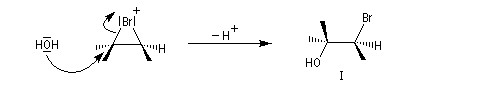
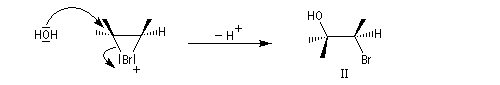
L'addition de BrOH sur le stéroïde ci-dessous est telle que le pont bromonium se forme du côté opposé au groupe méthyle. On obtient ainsi la molécule représentée et son énantiomère.
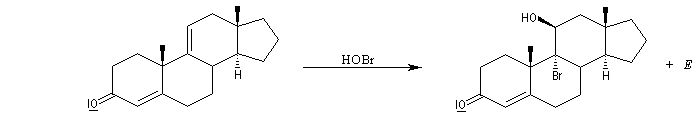
Utilisation du N-bromosuccinimide (NBS)
Le N-bromosuccinimide (NBS) réagit avec les composés éthyléniques en solvant aqueux pour donner des halohydrines.
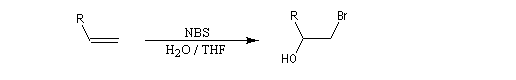
Oxymercuration-démercuration
Résultats expérimentaux
Lorsqu'on fait réagir un composé éthylénique avec l'acétate de mercure (II) dans un mélange d'eau et de THF et qu'on procède ensuite à la réduction du composé obtenu par le tétrahydroborate de sodium (borohydrure de sodium), on isole finalement un alcool [24], [30].
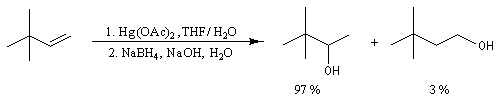
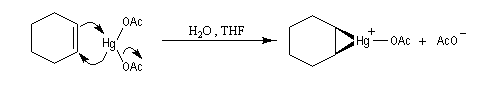
L'oxymercuration du cyclohexène fournit un mélange racémique de produits de stéréochimie trans. On interprète ce résultat par le mécanisme suivant :
- formation d'un ion mercurinium intermédiaire ;
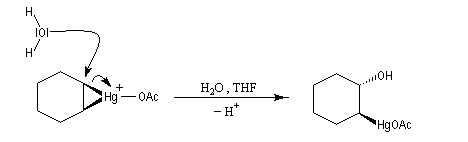
- ouverture de l'ion mercurinium par l'eau. Ce nucléophile réagit du côté opposé au pont.
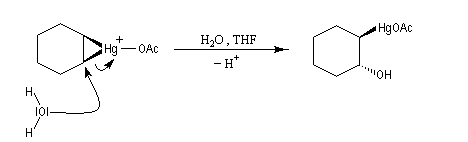
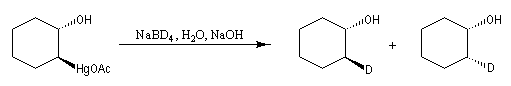
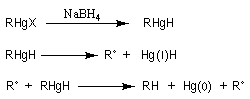

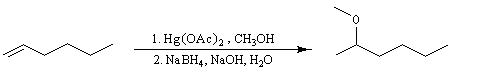
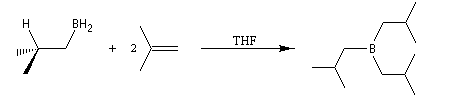
Résultats expérimentaux, préparation des organoboranes
L'hydratation acido-catalysée des composés éthyléniques fournit systématiquement l'alcool dont la classe est la plus élevée sur les deux possibles a priori car cette addition passe par l'intermédiaire du carbocation le plus stable. Pour obtenir l'alcool de la classe la moins élevée à partir d'un composé éthylénique, on peut opérer de façon indirecte en mettant à profit les propriétés remarquables des organoboranes. L'hydroboration des composés éthyléniques a été découverte en 1962 par le chimiste d'origine britannique H. C. Brown et son groupe lors de leurs recherches sur les propriétés réductrices des boranes.
Le monoborane BH3 est un composé déficient en électrons par rapport à l'octet. A la température ordinaire, il se dimérise pour conduire au diborane B2H6. Mais en présence de THF meilleure base de Lewis que H, il y a formation d'un complexe entre le monoborane et l'éther. Les solutions de BH3 dans le THF sont disponibles commercialement.
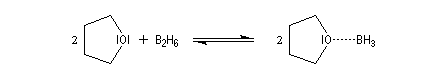
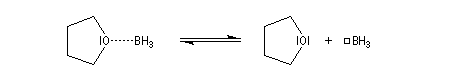
Avec un éthylénique peu substitué, une mole de BH3 réagit avec trois équivalents de composé éthylénique. On obtient un trialkylborane. On trouvera plusieurs exemples de ces réactions à la référence [17]. Il est amusant de constater que la suite H-C-B correspond aux initiales de H. C. Brown.
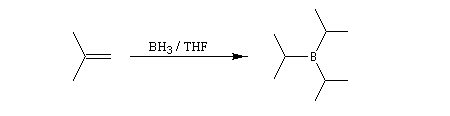
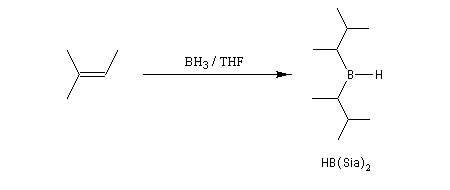
Sur le plan stéréochimique, on constate que l'addition est stéréospécifique de stéréochimie syn. Les composés éthyléniques plus substitués fournissent des dialkylboranes. Le Bis(1,2-diméthylpropyl)borane (appelé "disiamylborane", contraction de : di-secondary-isoamylborane) est un réactif très utile préparé généralement au moment de l'emploi par réaction entre le 2-méthylbut-2-ène et le complexe BH3-THF.
 |
Le diborane peut être obtenu par réduction du tétrahydroborate de sodium (borohydrure de sodium) par le trifluorure de bore dans un éther. A la température ordinaire c'est un gaz incolore. Il s'enflamme spontanément à l'air et réagit rapidement avec l'eau pour donner de l'hydrogène et de l'acide borique H3BO3. Sa structure comportant des atomes d'hydrogène en pont a été déterminée par diffraction électronique et spectroscopie Raman en 1951. Elle a suscité un énorme intérêt du fait du caractère original de ce type de liaison chimique. La structure des boranes peut être décrite en faisant intervenir des liaisons à 3 centres. Ce concept a été développé par le chimiste américain W. N. Lipscomb de l'Université de Harvard, prix Nobel de chimie 1976 [22]. |
Il s'agit d'une cycloaddition [2 + 2] entre une liaison bore-hydrogène et une liaison carbone-carbone. Le bore se fixe de façon régiosélective sur l'atome de carbone le moins substitué.
La régiosélectivité s'explique :
- par un facteur électronique car le bore est moins électronégatif que l'hydrogène ;
- par des facteurs stériques.

Un équivalent de borane réagit avec trois équivalents de composé éthylénique.
L'une des réactions les plus importantes des trialkylboranes est leur coupure oxydante par une solution de peroxyde d'hydrogène H2O2 en présence d'ions OH-.
L'oxydation d'un borane par une solution basique de peroxyde d'hydrogène fournit un alcool. Son importance provient du fait que le groupe OH se retrouve fixé sur l'atome de carbone le moins substitué du substrat. La régiosélectvité observée est donc l'inverse de celle obtenue dans une réaction d'hydratation en milieu acide qui suit la règle de Markovnikov.
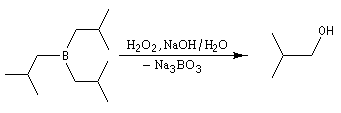
Cette réaction, et d'autres concernant la chimie du bore, ont valu à leur auteur H. C. Brown (Université de Purdue, Indiana) le prix Nobel de chimie en 1979 [21]. Le mécanisme comporte les étapes suivantes :
- formation de l'anion hydroperoxyde et réaction avec le trialkylborane ;
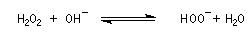
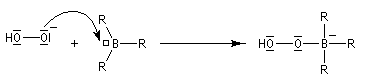
- transposition de l'intermédiaire organoborique. Sur le plan stéréochimique, le remplacement de la liaison C-B par une liaison C-O s'effectue avec rétention de la configuration ;
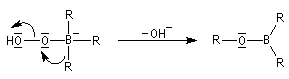
- la réaction conduit au remplacement des trois liaisons C-B par trois liaisons C-O successivement ;
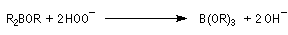
- le composé est finalement hydrolysé en alcool ;
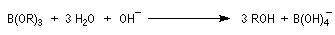
L'addition du diborane suivie de l'oxydation par H2O2 en milieu basique sur le méthylcyclohexène, fournit le mélange racémique des 2-méthylcyclohexanol de stéréochimie trans.
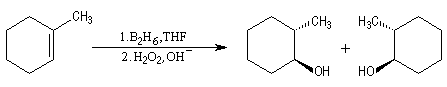
Utilisation du 9-BBN
Le 9-borabicyclo-[3, 3, 1]-nonane (9-BBN) est un agent d'hydroboration très utilisé en synthèse organique. On l'obtient en faisant réagir B2H6 avec le cycloocta-1, 5-diène [36].
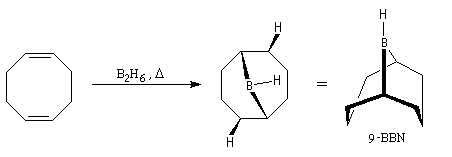
- contrairement à B2H6, il est stable à l'air ;
- alors que B2H6 réagit sans discernement avec la plupart des doubles liaisons éthyléniques, (9-BBN) est beaucoup plus chimiosélectif. Il réagit de façon préférentielle avec les doubles liaisons les moins encombrées et avec les composés cis de préférence aux trans.
 |
Le (+)-a-pinène représenté à gauche est un terpène qu'on trouve à l'état naturel dans l'essence de thérébentine. Il s'agit du (1R, 5R)-triméthyl-2, 6, 6-bicyclo[3, 1, 1]-hept-2-ène.
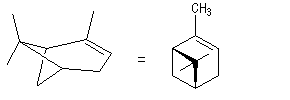 |
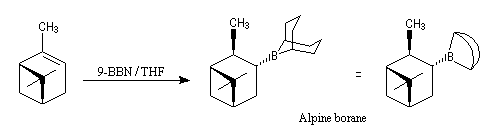
L'alpine borane est utilisé comme réducteur dans la réduction énantiosélective des composés carbonylés. Hydroboration énantiosélective
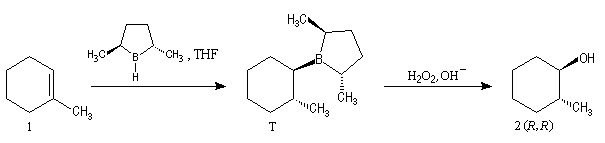
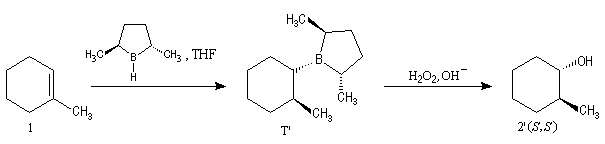
L'hydroboration du méthylcyclohexène par B2H6 suivie d'hydrolyse, fournit un mélange mélange racémique des 2-méthylcyclohexanols car le réactif achiral ne distingue pas les deux faces énantiotopiques du substrat.
Le premier réactif d'hydroboration énantiosélective a été préparé par H. C. Brown. Il s'agit du diisopinocamphéylborane Ipc2BH. On le prépare en faisant réagir B2H6 avec l' a-pinène. Selon l'énantiomère de l' a-pinène de départ, on obtient l'un ou l'autre des deux énantiomères du produit. L' excès énantiomérique est voisin de 100 %.
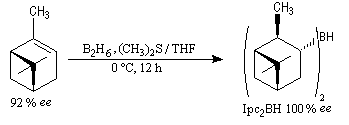
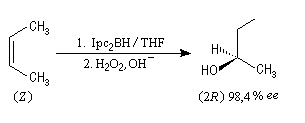
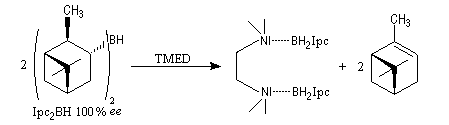
A cause de sa taille importante, Ipc2BH ne donne pas de bons résultats avec les alcènes trisubstitués. Pour effectuer l'hydroboration énantiosélective de ces composés on utilise de préférence le monoisocamphéylborane. Il donne aussi de très bons résultats avec les alcènes disubstitués trans. On peut préparer ce réactif au moyen de la réaction suivante (TMED désigne la tétraméthyléthylènediamine qui est un ligand bidenté).
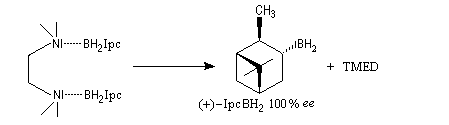
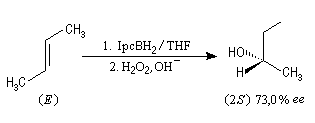
D'autres réactifs d'hydroboration ont été préparés à partir de composés éthyléniques chiraux comme le longifolène.
Additions radicalaires
Résultats expérimentaux
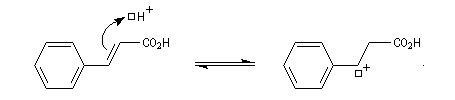
En étudiant l'addition de bromure d'hydrogène sous rayonnement ultraviolet le chimiste américain d'origine russe M. S. Kharasch a mis en évidence une addition de régiosélectivité inverse de celle observée pour l'addition ionique. L'orientation de l'addition est pour cette raison qualifiée d'anti-Markovnikov, terminologie qui se rapporte à la règle de Markovnikov. citée plus haut.
Mécanisme
Le mécanisme de cette réaction a été élucidé par M. S. Kharasch et F. R. Mayo en 1930. Il s'agit d'une réaction en chaîne.
- Initiation : elle peut être photochimique en utilisant un rayonnement de longueur d'onde convenable permettant
la coupure homolytique de la liaison de l'hydracide ;
ou être réalisée par voie thermique en présence d'un initiateur de radicaux ;
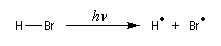
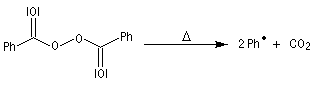
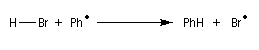
- Propagation : l'addition de l'hydracide s'effectue de manière à fournir le radical le plus stable, c'est à dire
celui qui est substitué par le plus grand nombre de groupes alkyles.
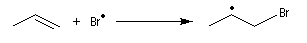
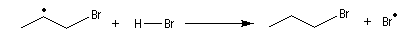
- Rupture : elle implique la recombinaison de radicaux et la présence d'une molécule du milieu permettant
d'emporter l'énergie excédentaire.
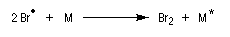
La régiosélectivité s'interprète par un contrôle cinétique de la réaction. L'étape cinétiquement déterminante est la formation du radical le plus stable.
Seul le bromure d'hydrogène est capable de réagir avec les composés éthyléniques selon ce mécanisme. En effet :
- avec HI, la première étape de propagation est très endothermique ;
- avec HCl c'est la deuxième étape de propagation qui est endothermique à cause de la stabilité assez élevée de la liaison entre l'hydrogène et le chlore.
Résultats expérimentaux
La découverte de la métathèse des alcènes a été faite en 1931. Il s'agissait de la disproportionation du propène en éthène et but-2-ène à haute température. Le premier exemple de métathèse catalysée a été rapporté par des chercheurs, R. L. Banks et G. C. Bailey en 1964 [57].
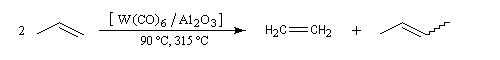
Les différents types de réactions de métathèse sont donnés ci-dessous.
- métathèse croisée (cross metathesis CM) ;
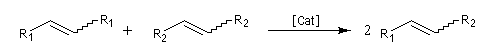
- fermeture de cycle (ring closing metathesis RCM) ;
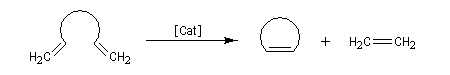
- polymérisation de diènes acycliques (acyclic dien metathesis polymerisation ADMEP) ;
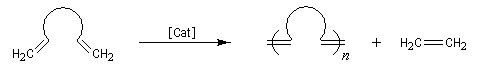
- ouverture de cycle (ring opening metathesis ROMP) ;
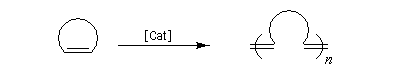
- métathèse des énynes (ring closing enyne metathesis RCEYM)
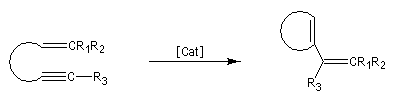
Le mécanisme de la métathèse des alcènes a été proposé par Y. Chauvin [58].
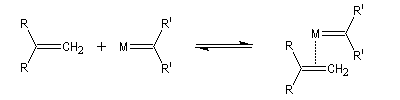

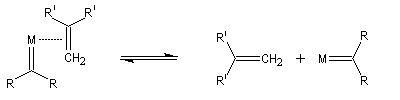
Les différentes étapes sont équilibrées. Pour rendre la transformation totale, on peut déplacer l'équilibre en jouant sur un facteur de celui-ci. L'effet entropique est favorable lorsqu'il y a production d'un gaz comme l'éthène.
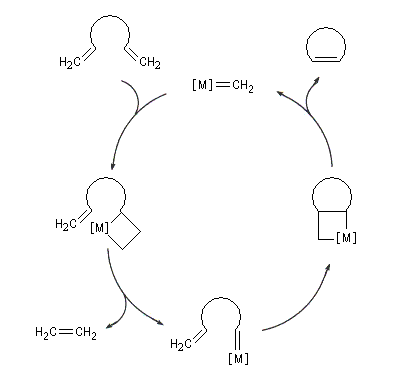
Le cycle catalytique est représenté ci-dessous dans le cas de la réaction de RCM d'un éthylénique possédant des groupes méthylènes terminaux. La force motrice de la réaction est la formation d'éthène.
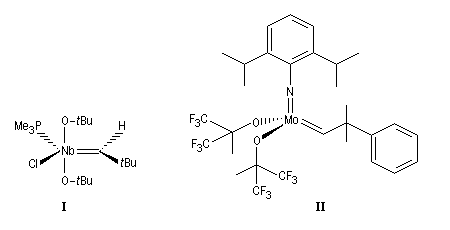
Le premier catalyseur réellement efficace en métathèse a été découvert par Schrock en 1980. Il s'agit d'un complexe métal-alkylidène au niobium représenté sur la figure ci-dessous (I). Le premier complexe métal-alkylidène avait lui même été préparé par Schrock en 1975. C'était un complexe du tantale. Schrock a ensuite préparé toute une série de catalyseurs au molybdène dont un exemple particulièrement efficace est (II).
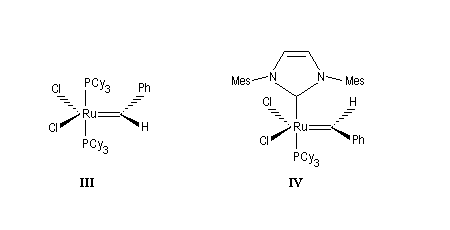
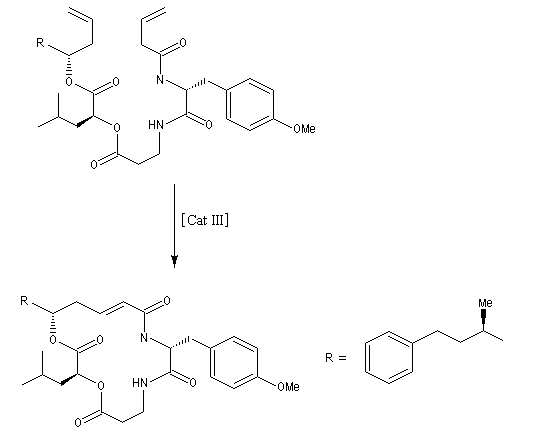
La réaction la plus importante pour les applications en synthèse organique est la réaction de cyclisation RCM qui peut être réalisée à l'aide du catalyseurs de Grubbs de première génération. Les fermetures de cycles à cinq chainons sont les plus faciles mais il est possible de synthétiser des cycles plus grands à partir de composés possédant des méthylènes terminaux. La métathèse croisée, plus difficile, nécessite des catalyseurs de deuxième génération.
La métathèse permet de réduire considérablement le nombre d'étapes dans la synthèse de molécules complexes. L'exemple suivant concerne la synthèse de l'arénastatine, un composé possédant des propriétés cytotoxiques. La synthèse de ce macrocycle peut également être effectuée par macrolactamisation.
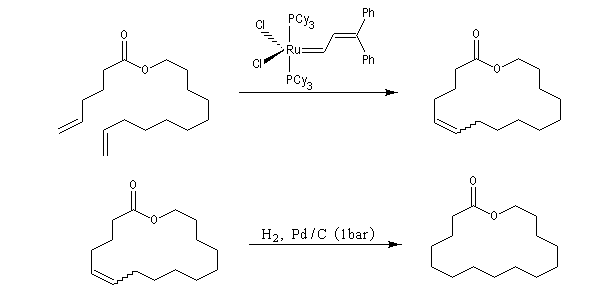
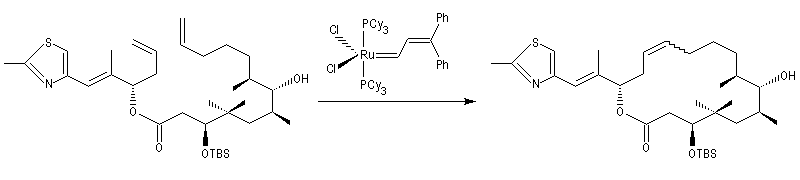
La métathèse du cyclooctatétraène est une très bonne méthode de préparation du polyacétylène.
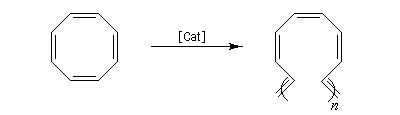
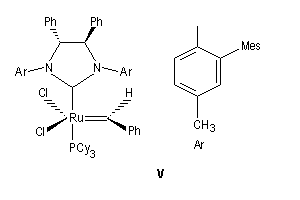
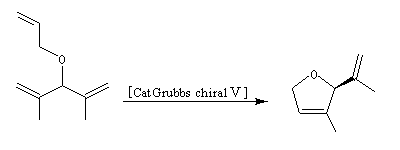
Le premier exemple de catalyseur chiral a été publié par Schrock en 1993. Il s'agissait d'un catalyseur au molybdène permettant une réaction de type ROMP énantiosélective. Le premier catalyseur au ruthénium pour la fermeture de cycle (V) a été publié par Grubbs en 2001.
Hydroformylation
Résultats expérimentaux
L'hydroformylation est une réaction industrielle découverte par le chimiste allemand O. Roelen en 1938. Le procédé porte aussi le nom de procédé oxo car il permet l'insertion d'oxygène dans une chaîne carbonée. La réaction fait intervenir un cycle catalytique assez complexe. Le bilan est le suivant :
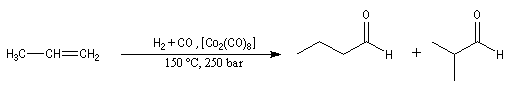
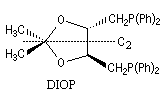
L'hydroformylation peut être réalisée de façon énantiosélective en utilisant un catalyseur soluble chiral du type de ceux rencontrés dans les réactions d'hydrogénation énantiosélective. La réaction suivante constitue une étape de la synthèse de la L-alanine. Le catalyseur est un complexe du rhodium comportant une diphosphine chirale appelée DIOP
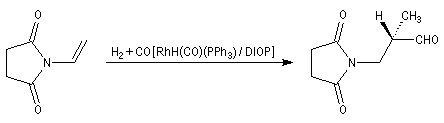
 |
 |
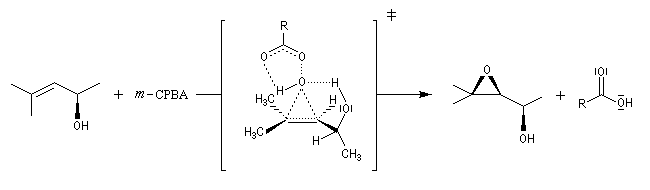
Préparation des époxydes La réaction entre un peroxyacide et un composé éthylénique permet la synthèse d'un époxyde (réaction de Prilezhaev, 1909). Plusieurs peroxyacides peuvent être utilisés, parmi lesquels : l'acide peroxybenzoïque, l'acide trifluoroperoxyacétique, l'acide métachloroperoxybenzoïque m-CPBA qui présente l'avantage d'être un solide qu'on peut conserver assez facilement. Le sous produit de la réaction est un acide carboxylique. L'utilisation d'un tampon basique permet d'éviter l'acidification de la solution. La réaction est généralement conduite dans le dioxane à une température proche de 0 °C. D'autres réactifs d'époxydation sont utiilisés :
- le monoperoxyphtalate de magnésium (MMPP) plus stable que m-CPBA ;
- le mélange oxydant suivant commercialisé par DuPont sous le nom d'oxone : 2 KHSO5 /KHSO4 / K2SO4 (notons que l'utilisation de ce réactif ne se limite pas aux époxydations des composés éthyléniques).
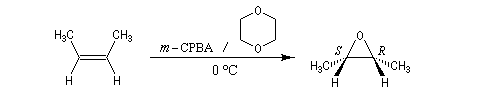
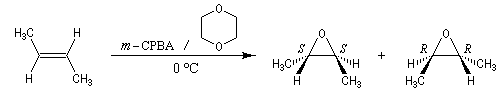
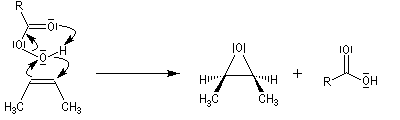
La réaction est stéréospécifique de stéréochimie syn. Cette stéréospécificité s'explique par le mécanisme ci dessous.
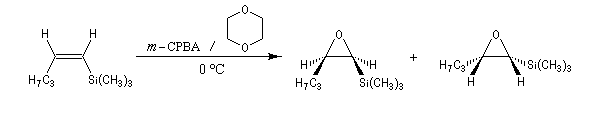
La réaction entre le m-CPBA et un vinylsilane fournit un époxysilane.
 |
L'acide métachloroperoxybenzoïque m-CPBA est très utilisé comme réactif d'oxydation en chimie organique. Il permet notamment l'époxydation des doubles liaisons éthyléniques. Comme tous les composés organiques comportant une liaison peroxo fragile, il est peu stable. Le composé pur est à l'origine de plusieurs accidents car il explose sous l'action d'un choc. Le réactif utilisé au laboratoire est constitué de 72 % de m-CPBA et de 10 % d'acide m-chlorobenzoïque. Il se présente sous la forme d'une poudre blanche qu'on doit conserver à basse température. Notons que ce réactif est également utilisé dans l'oxydation de Baeyer-Villiger des cétones, et dans la préparation des oxydes d'amines. Il est avantageusement remplacé par le monoperoxyphtalate de magnésium (MMPP). |
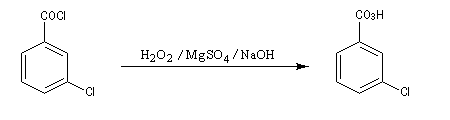
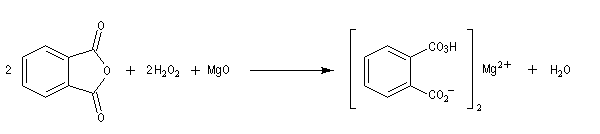
L'oxacyclopropane ou oxyde d'éthylène est obtenu dans l'industrie par réaction entre l'éthène, le dioxygène de l'air avec l'argent comme catalyseur (10 bar < P < 20 bar, T # 150 °C). C'est un intermédiaire important de la chimie industrielle. Il permet notamment la préparation de l'éthane-1,2-diol (éthylène glycol) utilisé dans la préparation des polyesters. Epoxydation diastéréosélective La tension allylique qui se développe dans la conformation 1 rend la conformation 2 plus stable que la précédente d'environ 12 kJ.mol-1.
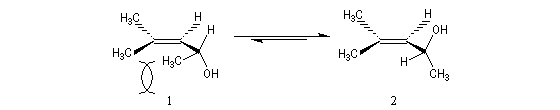
L'époxydation asymétrique des alcools allyliques a été mise au point par le chimiste américain K. B. Sharpless en 1980 [41]. Il s'agit d'un exemple de catalyse énantiosélective. Le substrat est un alcool allylique et l'oxydant est l'hydroperoxyde de tertiobutyle (TBHP). Ces composés sont tous les deux achiraux. La reconnaissance des faces énantiotopiques de la liaison éthylénique nécessite un catalyseur chiral. Celui-ci est un complexe entre l'ester diéthylique d'un acide tartrique chiral et le tétraisopropoxytitane. Puisque la réaction est catalytique, 5-10 % (mol) de Ti(OiPr4) en présence de tamis moléculaire 3 A suffisent.
L'exemple ci-dessous concerne la synthèse du (S)-glycidol (I), un époxyalcool utilisé dans la préparation de médicaments b-bloquants.
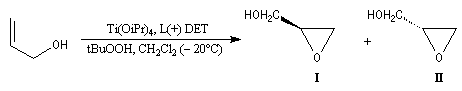
|
Composé
|
(S)-glycidol (I)
|
(R)-glycidol (II)
|
|
%
|
95
|
5
|
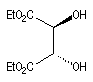 |
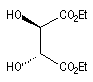 |
|
(2S, 3S)-D-(-)-DET
|
(2R, 3R)-L-(+)-DET
|
 |
L'image ci dessous représente la molécule de L-(+)-DET. 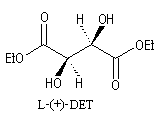 |
- l'époxydation a lieu sur la face supérieure avec le tartrate D-(-) ;
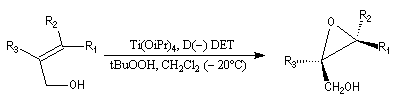
- elle a lieu sur la face inférieure (lower face) avec le tartrate L-(+).
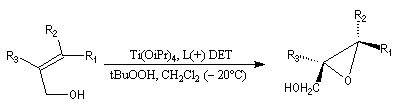
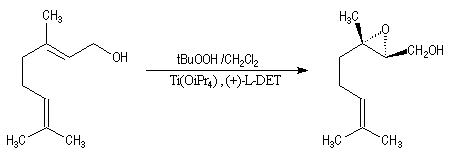
Peu de temps après sa découverte, l'époxydation de Sharpless a permis la synthèse d'une phéromone à l'échelle industrielle (J. T. Baker) : la disparlure ou (7R, 8S)-7, 8-époxy-2-méthyloctadécane. (phéromone de la mite gitane (gypsy moth). Les grandes lignes de sa synthèse sont données ci-dessous :
- l'étape clef de la synthèse est l'époxydation énantiosélective du substrat éthylénique de départ ;
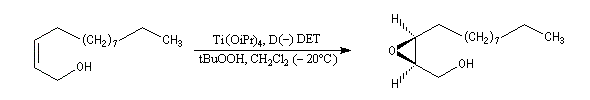
- l'oxydation de la fonction alcool primaire par le réactif de Collins permet l'arrêt au stade de l'aldéhyde ;
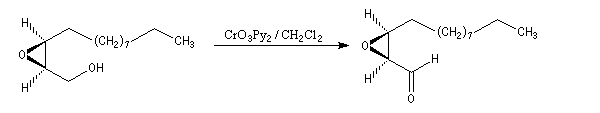
- la synthèse se poursuit par une oléfination de Wittig suivie d'une hydrogénation.
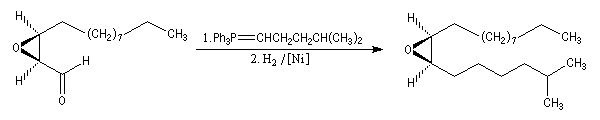
Le mécanisme exact de l'époxydation de Sharpless n'est pas connu avec certitude à l'heure actuelle. La présence d'une fonction alcool est essentielle pour que l'induction asymétrique ait lieu. K. B. Sharpless a obtenu le prix Nobel de chimie 2001 conjointement avec W. S. Knowles et R. Noyori [20].
Les époxydes dérivés d'alcools allyliques sont des 2,3-époxyalcools. Ils peuvent subir une réaction de transposition connue sous le nom de réarrangement de Payne.
Epoxydation énantiosélective de Jacobsen
Le substrat est un composé éthylénique de stéréochimie Z. L'oxydant (co-oxydant) est un hypochlorite. Le catalyseur est un complexe du Mn (III) utilisant un ligand chiral de symétrie C2 dérivé du ligand salen [47]. Ce réactif a été élu "réactif de l'année 1994". L'époxydation de Jacobsen est un exemple de catalyse énantiosélective.
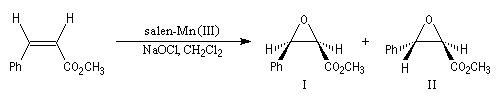
Elle est aussi énantiosélective. Pour l'exemple précédent, les excès énantiomériques sont donnés ci-dessous.
|
Composé
|
I
|
II
|
|
Excès énantiomérique (ee)
|
85
|
62
|
Dihydroxylation
Utilisation du permanganate de potassium
La syn-dihydroxylation vicinale des composés éthyléniques peut être effectuée en utilisant comme réactif une solution diluée et neutre de permanganate de potassium à 0 °C. Des expériences réalisées au moyen d'oxygène 18, ont permis de montrer que les atome d'oxygène du diol proviennent de l'ion permanganate. Cela suggère la formation d'un ester permanganique intermédiaire [55]. Des substrats diastéréo-isomères fournissent des produits diastéréo-isomères. La réaction est diastéréospécifique de stéréochimie syn. Cette stéréospécificité s'explique également par l'intervention de l'ester permanganique intermédiaire. Notons que l'état de transition présente une topologie aromatique à 6 électrons.
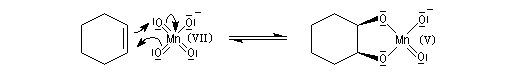
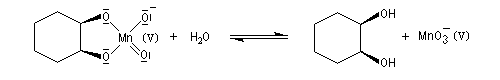
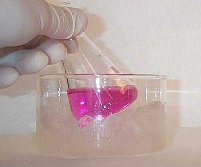 |
L'oxydation d'un composé éthylénique en diol par le permanganate de potassium doit être réalisée avec une solution diluée, en milieu neutre dans un bain de glace. Cette réaction constitue un test des composés éthyléniques appelé test de Baeyer. |
on en déduit l'équation de la réaction :
Lorsqu'on opère à la température ambiante, on observe une coupure oxydante de la chaîne carbonée et conduit à la formation de composés carbonylés. Les aldéhydes éventuels sont oxydés en acides carboxyliques.
La dihydroxylation diastéréospécifique anti des composés éthyléniques peut être effectuée en réalisant successivement la préparation d'un époxyde suivie de son ouverture par les ions hydroxydes.
Utilisation du tétroxyde d'osmium
Un réactif plus chimiosélectif que le permanganate et permettant d'effectuer la même transformation est le tétroxyde d'osmium OsO4. L'ajout d'une quantité stoechiométrique de tétroxyde d'osmium au composé éthylénique dans le THF conduit à un intermédiaire cyclique isolable. Là aussi, l'état de transition présente une topologie aromatique à 6 électrons.
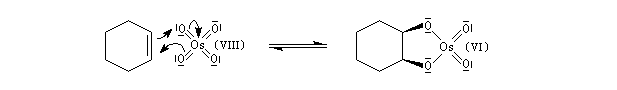
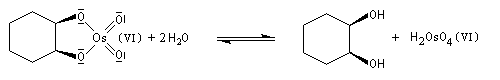
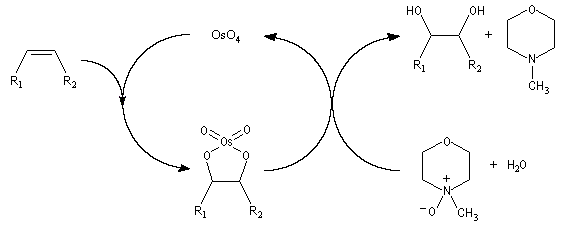
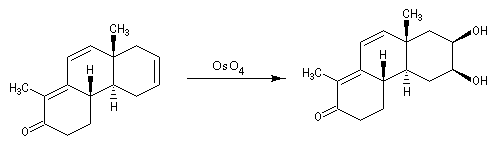

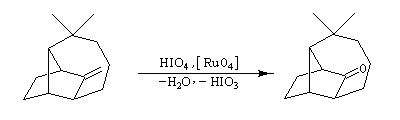
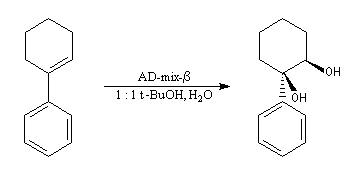
La dihydroxylation peut être rendue énantiosélective en utilisant une procédure mise au point par K. B. Sharpless en 1987 [41]. Comme l'époxydation énantiosélective du même auteur, il s'agit d'un exemple de catalyse énantiosélective. Mais alors que l'époxydation énantiosélective oblige à partir d'un alcène fonctionnalisé plus précisément un alcool allylique, la dihydroxylation peut être effectuée sur des liaisons éthyléniques ordinaires.
On trouve désormais dans le commerce des mélanges prêts à l'emploi appelés AD-mix-a et AD-mix-b. Leur composition est donnée ci-dessous :
|
Catalyseur
|
AD-mix-a
|
AD-mix-b
|
|
composition
|
1,4-bis(9-O-dihydroxyquinidine)phtalazine (DHQ)
K2OsO5; K3Fe(CN)6 ; NaHCO3 ; MeSO2NH2 |
1,4-bis(9-O-dihydroxyyquinidine)phtalazine (DHQD)2-PHAL
K2OsO5; K3Fe(CN)6 ; NaHCO3 ; MeSO2NH2 |
RG > RM > RP
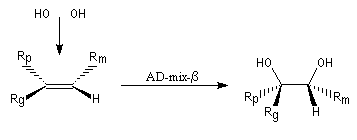
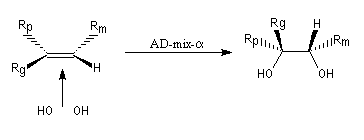
|
élément chiral
|
élément chiral
|
motif central
|
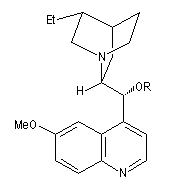 |
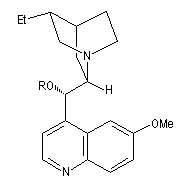 |
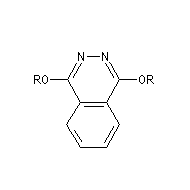 |
|
dihydroquinine (DHQ)
|
dihydroquinidine (DHQD)
|
phtalazine (PHAL)
|
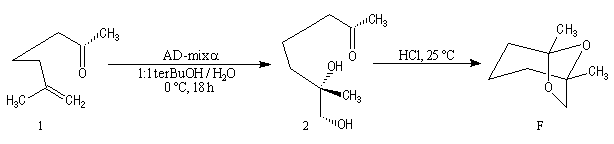
Aminohydroxylation énantiosélective (AA)
L'aminohydroxylation des composés éthyléniques concernait à l'origine la synthèse d'un mélange racémique d'aminoalcools en utilisant la chloramine T (Na+ClN- Ts) comme réactif [46], [62]. La version énantiosélective par catalyse asymétrique a été décrite par K. B. Sharpless en 1996 [48]. Elle fait partie de la trilogie des réactions énantiosélectives (EA, AD, AA) qui ont valu le prix Nobel de chimie 2001 à leur auteur.
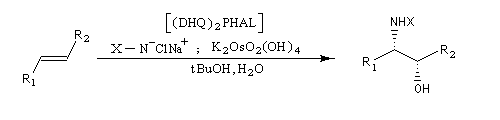
Synthèses d'oxazolidinones utilisées comme auxilliaires chiraux dans l'alkylation diastéréosélective d'Evans.
La synthèse suivante, au cours de laquelle l'intermédiaire n'est pas isolé s'effectue en une seule fois dans le même réacteur. C'est un exemple de réaction monotope (mono : unique, topos : endroit ; en anglais : one pot reaction.)
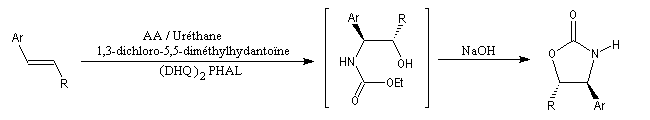
Ar = Ph, 4-MeO, 4-O2NC6H5.
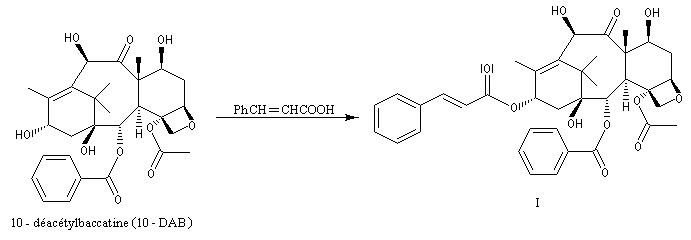
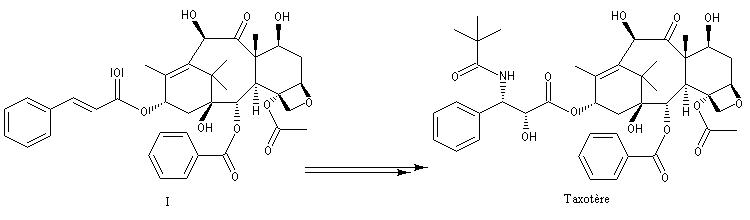
Peu de temps après sa découverte, la réaction d'aminohydroxylation énantiosélective a trouvé une application dans une synthèse élégante de la chaîne latérale du taxol en seulement quatre étapes [49].
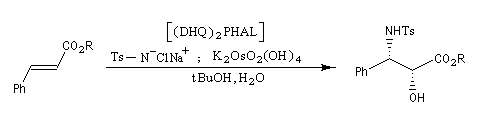
.
Résultats expérimentaux
Le trioxygène ou ozone est un allotrope du dioxygène. L'ozone donne des réactions intéressantes avec les composés éthyléniques. Celles-ci peuvent servir de méthode préparatives des composés carbonylés. Elles ont aussi été utilisées pour préciser la position d'une double liaison dans une molécule.
 |
Le chimiste allemand C. F. Shönbein découvrit le trioxygène ou ozone O3 en 1839. L'ozone est présent à l'état naturel dans la haute atmosphère de la terre. L'étude de la formation et de la décomposition de l'ozone a été récompensée par l'attribution du prix Nobel de chimie en 1995 à P. J. Crutzen, M. J. Molina et F. Sherwood Rowland [60]. Au laboratoire on le prépare en faisant traverser le dioxygène O2 par une effluve électrique dans un ozoniseur [53]. A la température ordinaire l'ozone est un gaz de couleur bleue possédant une odeur piquante qui lui a valu son nom (ozein signifie odeur en grec). L'ozone se liquéfie à -112 °C pour donner un liquide bleu explosif. Conformément au prévisions de la méthode VSEPR, la molécule d'ozone a la forme d'un V avec un angle entre les liaisons de 116,8°. |
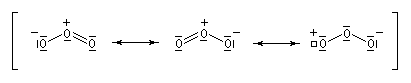
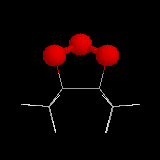 |
L'image de gauche représente l'ozonide initial formé dans le cas de la cycloaddition de l'ozone sur le 1,1,2,2-tétraméthyléthène.
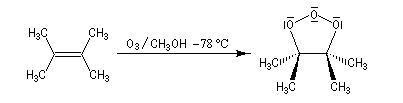 |
Le mécanisme actuellement admis a été proposé par R. Criegee en 1951. La réaction entre O3 et la double liaison éthylénique est une cycloaddition dipolaire 1, 3. Le produit d'addition s'appelle ozonide initial ou molozonide. Cette cycloaddition peut être interprétée en utilisant l'approximation des orbitales frontières. L'état de transition présente une topologie aromatique à 6 électrons.
Formellement, la molécule d'ozone ressemble beaucoup à un anion allylique. Les orbitales moléculaires p de l'éthylène ont été décrites plus haut.
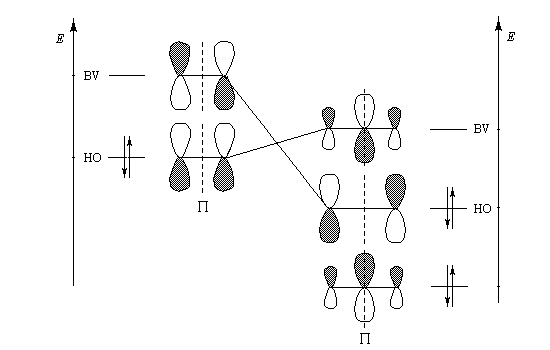
- formation de l'ozonide initial (molozonide) par cycloaddition dipolaire 1,3 ;
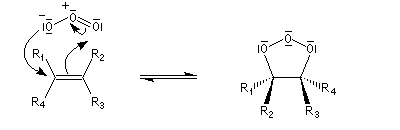
- l'ozonide initial est particulièrement instable du fait de
l'existence de deux liaisons peroxo successives. Il se fragmente
spontanément au cours d'une réaction de rétro-cycloaddition ;
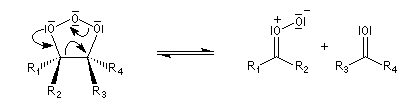
- formation de l'ozonide secondaire.
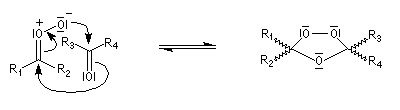
Ozonolyse
L'une des principales réactions des ozonides est leur coupure en composés carbonylés. Celle-ci peut être effectuée sous l'action de plusieurs réactifs :
- le zinc dans l'acide éthanoïque réduit les ozonides en aldéhydes ou cétones ;
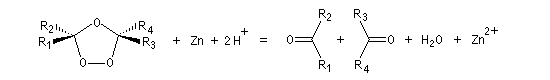
- une procédure plus moderne consiste à utiliser le diméthylsulfure (CH3)2S qui est alors oxydé en diméthylsulfoxyde (DMSO).
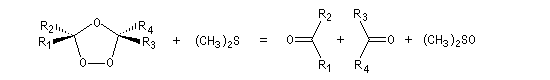
- l'hydrolyse en milieu non réducteur (ozonolyse oxydante)
fournit du peroxyde d'hydrogène comme sous-produit. Ce dernier provoque
l'oxydation les aldéhydes éventuellement formés en acides carboxyliques.
L'ozonolyse oxydante est peu utilisée en synthèse organique. On préfère en général travailler en milieu réducteur, isoler les produits quitte à les oxyder ensuite.
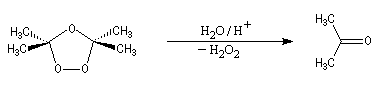
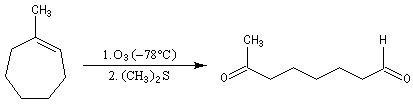
Depuis sa découverte par C. D. Harries en 1903, la réaction a été largement utilisée pour localiser la position des doubles éthyléniques dans les molécules organiques.
Supposons que l'ozonolyse d'un alcène fournisse uniquement de la butanone et du méthanal. On en déduit qu'il s'agit du 2-méthylbut-1-ène.
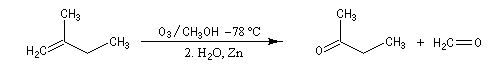
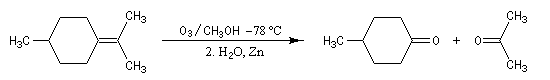
L'ozonolyse est aussi utilisée pour préparer des composés carbonylés ou dicarbonylés. Une préparation des composés dicarbonylés 1,4 consiste à effectuer l'ozonolyse d'un cyclobutène disubstitué préparé préalablement, par exemple, en effectuant une cycloaddition entre un cétène et un composé éthylénique.
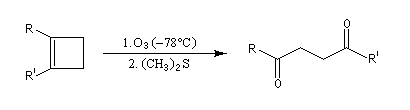
- addition d'un dithiane lithié sur un accepteur de Michaël ;
- hydrolyse en milieu acide de dérivés disubstitués du furane ;
- Addition de Michaël d'un carbanion préparé à partir d'un dérivé nitré sur une a-énone, suivie d'une réaction de Nef.
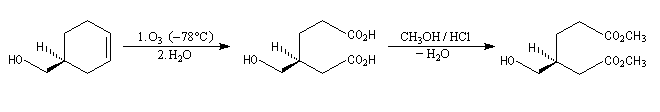
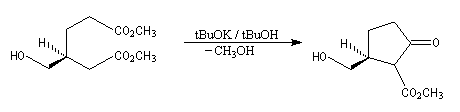
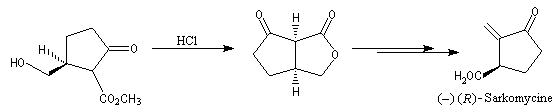
Oxydation ménagée catalysée par le palladium
Oxydation en aldéhyde
L'oxydation de l'éthylène par le dioxygène de l'air permet la préparation industrielle de l'éthanal. Le procédé, mis au point en 1953 porte le nom de procédé Wacker. Il a supplanté la préparation de l'éthanal par hydratation de l'éthyne (acétylène). La réaction fait intervenir un cycle catalytique complexe mettant en jeu des composés organopalladés. Les principales étapes sont les suivantes :
- oxydation de l'éthylène par le palladium (II) et réduction concomittante de ce dernier en Pd (0) ;
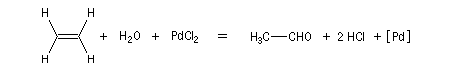
- oxydation du Pd (0) en Pd (II) par le chlorure de cuivre (II) avec réduction de ce dernier en chlorure de cuivre (I) ;

- oxydation du chlorure de cuivre (I) en chlorure de cuivre (II) par le dioxygène de l'air.
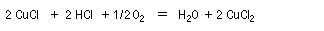
La réaction d'oxydation des composés éthyléniques catalysée par le chlorure de palladium constitue une méthode générale de préparation des méthylcétones à partir des alcènes terminaux. Comme les méthyl cétones sont des intermédiaires importants en synthèse organique, la réaction possède un indéniable intérêt pratique. Cette méthode est plus intéressante que l'alkylation de l'acétone qui souffre d'un manque de sélectivité [25].
Coupure oxydante
Lorsqu'on utilise une solution concentrée de permanganate de potassium, à chaud, on observe une coupure oxydante de la double liaison éthylénique. Le trioxyde de chrome est également utilisé. Les composés éthyléniques disubstitués donnent des aldéhydes qui sont suroxydés immédiatement en acides carboxyliques.
D'où l'équation de réaction :
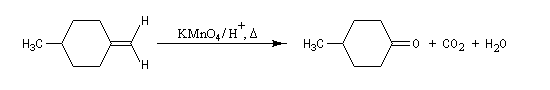
Les composés éthyléniques tétrasubstitués donnent des cétones.
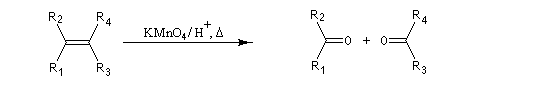
Transposition de Cope
La transposition de Cope une réaction qui transpose le squelette des diènes-1,5. Du point de vue mécanistique, il s'agit d'une transposition sigmatropique [3,3]. Si le diène n'est pas substitué, le produit d'arrivée est le même que le substrat de départ. La réaction est dégénérée. Cette transposition se rapproche de celle des éthers allyliques dite transposition de Claisen.
Bibliographie
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Miller, Neuzil, Modern Experimental Organic Chemistry (DC Heath, 1982).
Liens
[13] Than Tung Nguyen Dang - Eléments de chimie quantique
[14] Wilkinson catalyst
[15]
[16] Cholest-5-ene-3-one by L. F. Fieser Harvard University
[17] Ketones and Alcohols from organoboranes by Hiromichi Kono and John Hooz University of Alberta
[18] Sharpless Dihydroxylation
[19] Enantioselective Epoxydation of Allylic Alcohols
[20] The Nobel Prize in Chemistry 2001
[21] The Nobel Prize in Chemistry 1979
[22] The Nobel Prize in Chemistry 1976
[23] Assymetric hydrogenation of allylic alcohols using BINAP-ruthenium complexes by H. Takaya, T. Ohta, S. Inoue, M. Tokunaga and R. Noyori.
[24] Oxymercuration-reduction. Alcohols from olefins, 1-méthylcyclohexanol by J. M. Jerkunica and T. G. Traylor
[25] A general synthetic method for the preparation of methyl ketones from terminal olefins : 2-decanone by Jiro Tsuji, Hideo Nagashima, and Hisao Nemoto.
[26] Aldehydes from olefins
[27] Bromohydrins from alkenes and NBS by A. W. Langman and D. R. Dalton.
[28] (+)-isopinocampheol, a simple and convenient method for oxidation of organoboranes using sodium perborate by George W. Kabalka, John T. Maddox, Timothy Shoup, and Karla R. Bowers.
[29] Hydroboration by Jonathan Goodman, Cambridge University Chemical Laboratory.
[32] Studies toward new reagents for asymetric hydroboration by Kevin J. Hodgetts, Gerhard Lascober and Massimo Zorzi.
[36] 9-borabicyclo[3, 3, 1]nonane dimer by John A. Soderquist and Alvin Negron.
[37] Stereocontrolled Iodolactonization of Acyclic Olefinic Acid by F. Bermejo Gonzalez and Paul A. Bartlett.
[38] Sharpless dihydroxylation
[39] The Nobel Prize in Chemistry 1912
[40] (1S,2R) and (1R,2S)-trans-2-phenylcyclohexanol by Sharpless Asymmetric Dihydroxylation by Javier Gonzalez, Christine Aurigemma, and Larry Truesdale
[41] Searching for New Reactivity, Nobel Lecture, December 8, 2001, by K. Barry Sharpless, The Scripps Research Institute
[42] Thèse Delacroix
[43] Thèse Nury
[44] Sharpless aminohydroxylation
[50] The Sharpless epoxidation of allylic alcohols
[51] Asymmetric aminohydroxylation of alkenes
[52] Nobel Lectures H. C. Brown
[53] Ozone by L. I. Smith, F. L. Greenwood, and O. Hudrlik
[54] Ozonolytic cleavage of cyclohexene by Ronald E. Claus and Stuart L. Schreiber
[60] The Nobel Prize in Chemistry 1995
[61] The Nobel Prize in Chemistry 1994
[62] Osmium-Catalyzed Vicinal Oxiamination by Chloramine T by Eugenio Herranz and K. Barry Sharpless
[63] Grubbs reagent
[64] E. J. Corey, The Nobel Prize in Chemistry 1990
[65] Spectres ABC des alcènes
[66] Cohalogenation of Limonen
[67] Ring-Closing Metathesis Synthesis of N-Boc-3-pyrrolin by Marcelle L. Ferguson, Daniel J. O'Leary , and Robert H. Grubbs
[68] Metathesis – a change-your-partners dance.
[69] Les porphyrines, structure propriétés et applications.
[70] La méthathèse : de Chauvin à la chimie verte
[71] Epothilones
[72] Orbitales frontières, cycloadditions Cours de L. Grimaud, ENSTA.
[73] Thèse Royer, Ecole Polytechnique
[74] Introduction à la chimie moléculaire par les orbitales frontières Pascal le Floch, Ecole Poytechnique.
[75] Assymetric Hydrogenations, by W. S. Knowles.
Articles
[30] R. Gibbs and W. P. Weber, J. Chem. Educ. 48 (1971), 477.
[31] W. S. Knowles, J. Chem. Educ. 63 (191986), 222-225.
[33] Brown, H.C. ; Yoon, N. M. J. Am. Chem. Soc., 1977, 99, 5514.
[34] Brown, H.C. ; Zweifel, G. J. Am. Chem. Soc., 1961, 83, 486
[35] Masamune, S.; Kim, B -M. ; Petersen, J. S. ; Sato, T. ; Veenstra, S. J. J. Am. Chem. Soc., 1985, 107, 4549.
[45] E. N. Jacobsen et al., J. Am. Chem. Soc.110, 1968 (1988).
[46] K. B. Sharpless et al., J. Am. Chem. Soc. 97, 2305 (1975)
[47] E. N. Jacobsen et al. J. Am. Chem. Soc 113, 7063 (1991).
[48] G. Li, H. T. Chang, K. B. Sharpless, Angew. Chem. Int. Ed. Engl., 1996, 35, 451.
[49] G. Li, K. B. Sharpless, Acta Chem. Scand 1996, 35, 451.
[55] K. Wiberg et K. Saegebarth, The Mechanisms of Permanganate Oxidation J. Am. Chem. Soc.79, 2822 (1957).
[56] C. Djerassi and C. R. Scholz, J. Am. Chem. Soc.70, 417 (1948).
[57] R. L. Banks and G. C. Bailey, Ind. Eng. Chem., Prod. Res. Develop., 3; 170, (1964).
[58] J. L. Hérisson et Y. Chauvin, Makromol. Chem., 141, 161 (1970).
[59] Robert K. Boeckman Jr., Paul C. Naegely, and Samuel D. Arthur. J. Org. Chem., 1980
[80] R. B. Woodward, F. Sondheimer, D. Taub, K. Heusler and W. M. McLamore J. Amer. Chem. Soc. 74, 4223 (1952).
[81] A. C. Cope, E. M. Hardy, J. Am. Chem. Soc. 1940, 62, 441.
[82] Produits naturels anticancéreux, Daniel Guénard, Françoise Guéritte, Pierre Potier, l'actualité chimique, avril-mai 2003.
[83] Vineyard, B.D.; Knowles, W.S.; Sabacky, M.J.; Bachman, G.L.; Weinkauff, D.J. J. Am.Chem. Soc. 1977, 99, 5946. “Asymmetric Hydrogenation. Rhodium Chiral Bisphosphine Catalyst”
Ouvrages théoriques
A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
J. March - Advanced organic chemistry, Wiley Interscience.
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
V. Minkine - Théorie de la structure moléculaire, Editions Mir 1979.
N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
P. Atkins - Molecular Quantum Mechanics, Oxford University Press 1983.
J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.









0 commentaires:
Enregistrer un commentaire