Cours de chimie Organique -
Dérivés halogénés et substrats apparentés
Introduction
Nomenclature
Les dérivés halogénés des alcanes ou halogénures d'alkyles sont des composés qui dérivent formellement des hydrocarbures par remplacement d'un atome d'hydrogène par un atome d'halogène.
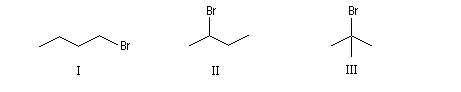
|
I
|
II
|
III
|
|
1-Bromobutane
|
2-Bromobutane
|
2-Méthyl-2-bromopropane
|
Il existe des dérivés halogénés en série alicyclique ainsi que des composés dans lesquels l'halogène est relié à un groupe vinyle ou allyle.
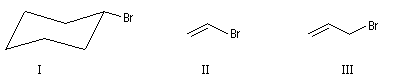
|
I
|
II
|
III
|
|
Bromocyclohexane
|
2-Bromoéthène
|
3-bromopropène
|
Les dérivés halogénés sont des agents cancérogènes du fait de leur caractère alkylant. Les dérivés méthylés sont particulièrement toxiques et ne doivent être manipulés qu'avec beaucoup de précautions. Classe
Selon que l'atome de carbone portant l'halogène est lié à 1, 2, 3 atomes de carbone, le dérivé est qualifié de primaire, secondaire, tertiaire.
|
Dérivé halogéné
|
Bromo-1-butane
|
Bromo-2-butane
|
2-Méthyl-2-bromopropane
|
|
Classe
|
I
|
II
|
III
|
Etat naturel
Pourpre
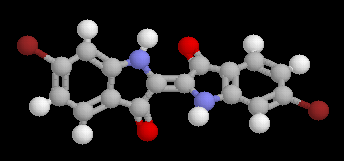 |
Le pourpre qui ornait le bas de la toge romaine est un composé bromé extrait du murex, un coquillage abondant en Méditérannée. Ce colorant d'origine naturelle est connu depuis très longtemps puisque des textes en mentionnent l'existence en 1600 avant J.-C. La première synthèse de ce composé a été réalisée par Sachs et Kempf en 1903. On trouvera des informations intéressantes sur ce composé à la référence [20]. |
Propriétés physiques
Caractéristiques physiques et énergétiques
|
Elément
|
C
|
F
|
Cl
|
Br
|
I
|
|
Electronégativité (Pauling)
|
2,55
|
3,98
|
3,16
|
2,96
|
2,66
|
Le tableau suivant regroupe les températures de changement d'état des halogénures de méthyle.
On constate une augmentation du moment dipolaire lorsqu'on passe du dérivé iodé au dérivé fluoré. Cet accroissement est à mettre en relation avec l'augmentation de l'électronégativité de l'halogène. Cependant il faut faire attention que le moment dipolaire fait intervenir la longueur de liaison qui varie aussi.
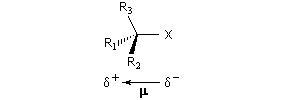
|
Composé
|
CH3-H
|
CH3-F
|
CH3-Cl
|
CH3-Br
|
CH3-I
|
|
TE (°C)
|
-164
|
-78
|
-24
|
3
|
42
|
|
Longueur C-X (pm)
|
101
|
135
|
177
|
194
|
214
|
|
Do (kJ.mol-1)
|
435
|
485
|
327
|
285
|
213
|
|
Moment dipolaire m (D)
|
0
|
1,92
|
2,05
|
2,01
|
1,87
|
- dipôle permanent dipôle induit ;
- dipôle instantané dipôle induit.
Spectroscopie infrarouge
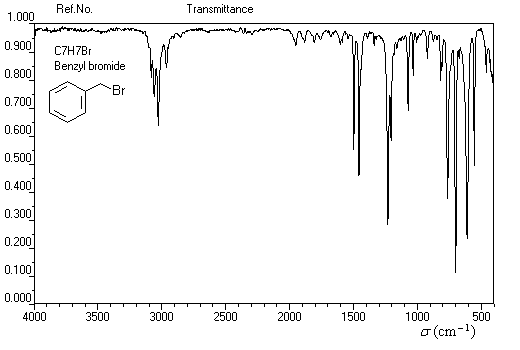
Dans les composés suivants, on observe une augmentation du déplacement chimique avec l'accroissement de l'électronégativité de l'halogène. Ce phénomène est général. Plus l'halogène est l'électronégatif plus la densité électronique autour du noyau d'hydrogène est faible. L'intensité du champ nécessaire pour atteindre la résonance diminue. Il en résulte un accroissement du déplacement chimique.
|
Composé
|
CH3I
|
CH3Br
|
CH3Cl
|
CH3F
|
|
Déplacement chimique d (ppm)
|
2,3
|
2,7
|
3,4
|
4,5
|
Définitions
Nous allons étudier des réactions de substitution c'est à dire des réactions au cours desquelles on observe la rupture d'une liaison et la reformation simultanée ou non d'une autre liaison.
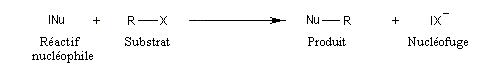
Dans le cas qui nous occupe ici, le substrat est un halogénure d'alkyle. Le réactif est un nucléophile (étymologiquement : qui a une affinité pour les noyaux ; c'est à dire les zones déficientes en électrons). Ce terme se réfère à l'aptitude que possède une base de Lewis à céder son doublet. On peut résumer cela de façon formelle formelle par l'équation ci-dessous.
Les réactions de substitutions nucléophiles sont notées SN selon la terminologie d'Ingold. Il existe une assez grande variété de mécanismes permettant de rendre compte des réactions de substitution nucléophile et l'on observe une certaine continuité entre-eux. Cependant nous allons voir qu'on peut dégager deux mécanismes limites pour ces réactions :
- la rupture de la liaison C-X et la formation de la liaison C-Nu ont lieu de façon concomitante (on dit aussi parfois de façon concertée, de l'anglais : concerted) ;
- la rupture de la liaison C-X précède la formation de la liaison C-Nu.
Remarque : les dérivés halogénés aromatiques ou vinyliques n'ont pas du tout la même réactivité que les halogénures d'alkyle dans les réactions de substitutions nucléophiles. Ils ne réagissent pas dans les conditions ordinaires. Ils donnent lieu à des réactions de couplage catalysées par des métaux de transition comme le palladium.
Substitutions nucléophiles bimoléculaires
Résultats expérimentaux, inversion de Walden
La réaction entre les ions iodure et (2S)-2-bromopropane, fournit le (2R)-2-iodopropane . La réaction s'accompagne d'une inversion de la configuration relative du centre chiral appelée inversion de Walden.
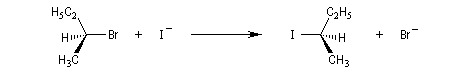
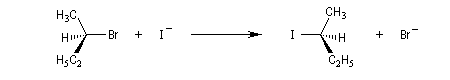
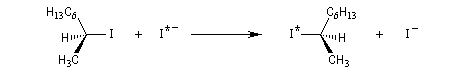
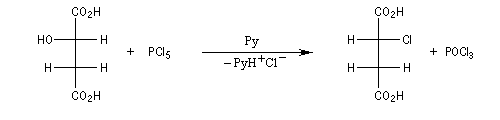
Mécanisme et cinétique
Le mécanisme admis est un choc bimoléculaire entre le nucléophile et le substrat. Il s'agit d'une réaction élémentaire.
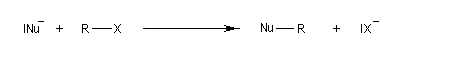
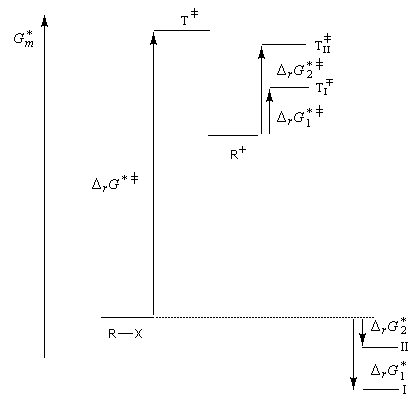
Puisque la transformation s'effectue en une seule étape élémentaire, le profil énergétique comporte un seul état de transition qui traduit la rupture partielle de la liaison entre l'atome de carbone et le nucléofuge et la formation partielle concomitante de la liaison entre l'atome de carbone et le nucléophile.
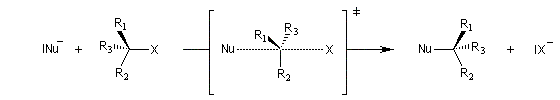
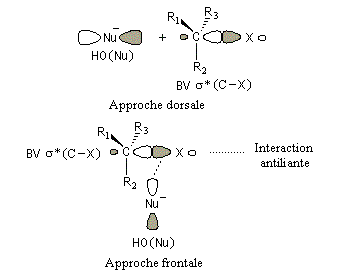 |
Interprétation orbitalaire de l'inversion de Walden. Dans l'approximation des orbitales frontières, l'interaction principale a lieu entre la plus haute orbitale occupée du nucléophile (HO) et la
plus basse orbitale vacante du substrat (BV) qui est l'orbitale antiliante de la liaison C-X.
|
Cet aspect est étudié dans un chapitre particulier consacré aux bases de Lewis.
Nature du nucléofuge
|
Liaison
|
C-C
|
C-F
|
C-Cl
|
C-Br
|
C-I
|
|
Vitesse relative
|
-
|
10-4
|
2,0´10-2
|
1,0
|
3,0
|
|
Do (kJ.mol-1)
|
-
|
485
|
340
|
285
|
213
|
|
Polarisabilité 1030´a (m-3)
|
0,5
|
0,7
|
2,5
|
3,6
|
5,6
|
|
Nucléofugacité
|
-
|
1
|
2,0´102
|
104
|
3,0´104
|
Commençons par examiner l'influence de l'encombrement de l'atome de carbone en a de l'halogène (solvant : propanone à 298 K).
|
Composé
|
CH3Br
|
CH3CH2Br
|
(CH3)2CHBr
|
(CH3)3CBr
|
|
Vitesse relative
|
145
|
1,0
|
7,7´10-3
|
4,0´10-6
|
 |
 |
 |
|
CH3CH2Br
|
(CH3)2CHBr
|
(CH3)3CBr
|
|
Composé
|
CH3CH2Br
|
CH3(CH2)2Br
|
(CH3)2CHCH2Br
|
(CH3)3CCH2Br
|
|
Vitesse relative
|
1
|
0,8
|
3,0´10-3
|
1,3´10-5
|
|
Dérivé halogéné
|
tBuCH2Br
|
tBu(CH2)2Br
|
tBu(CH2)3Br
|
|
Vitesse relative
|
1,3´10-5
|
3´10-2
|
1
|
![1-bromobicyclo[2,2,2]octane](http://www.faidherbe.org/site/cours/dupuis/images24/quinubr.gif) |
La figure de gauche représente le 1-bromobicyclo[2,2,2]octane. Ce composé n'est pas du tout réactif dans les réactions de substitutions nucléophiles SN2 à cause de l'encombrement stérique de l'atome de carbone porteur de l'halogène. Il ne réagit pas non plus selon un processus SN1 car cela impliquerait la formation d'un carbocation non plan. |
On s'intéresse à la réaction suivante pour différents groupes R (solvant : acétone).
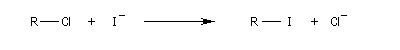
|
Composé
|
CH3(CH2)2Cl
|
CH2=CHCH2Cl
|
PhCH2Cl
|
CH3COCH2Cl
|
|
Vitesse relative
|
1,0
|
75
|
180
|
33 000
|
Notons qu'à du mécanisme SN2 ordinaire, il existe un mécanisme particulier nommé SN2' observé chez les systèmes allyliques.
Les composés carbonylés a-halogénés réagissent rapidement. La mobilité de l'halogène est accrue par l'interaction entre le nucléophile et le groupe carbonyle. Leur hydrolyse rapide dans les yeux provoque un comportement lacrymogène.
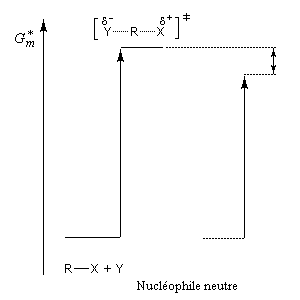
Influence du solvant
L'influence qualitative de la nature du solvant sur les substitutions nucléophiles a été étudiée par Hughes et Ingold. Le modèle consiste à comparer la solvatation des réactifs avec celle de état de transition. Nous nous limitons ici aux réactions SN2 faisant intervenir un dérivé halogéné comme substrat. Celui-ci est donc neutre. La théorie de Hughes et Ingold fait intervenir seulement des interactions de nature électrostatique. Plus la solvatation d'une espèce par le solvant est grande, plus l'énergie de la nouvelle entité solvatée est abaissée. D'autre part, une espèce est d'autant plus solvatée que sa charge électrique est plus élevée et concentrée dans un volume plus petit.
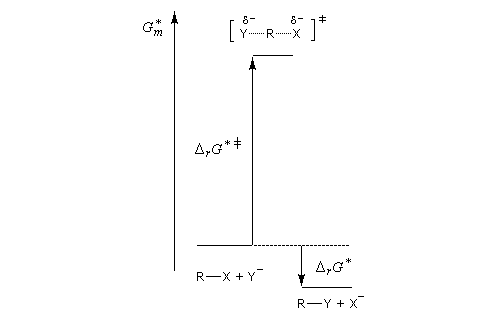
Puisque l'énergie d'activation est la différence entre l'énergie de l'état de transition et celle des réactifs, on en déduit :
- si les réactifs sont moins solvatés que l'état de transition, l'énergie d'activation diminue et on observe une accélération ;
- si les réactifs sont plus solvatés que l'état de transition, l'énergie d'activation augmente et on observe une diminution de vitesse.
 |
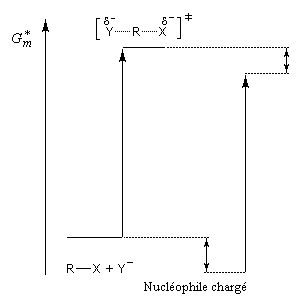 |
|
Création de charges contraires dans l'état de transition. Augmentation de la vitesse
|
Charge plus dispersée dans l'état de transition que dans les réactifs. Diminution de la vitesse
|
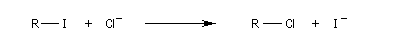
I- > Br- > Cl-
|
Solvant
|
Permittivité relative er
|
Moment dipolaire
|
|
38
|
3,8
|
|
|
49
|
4,3
|
|
|
30
|
5,5
|
Applications diverses
Nous allons décrire dans ce paragraphe quelques exemples d'alkylation de réactifs inorganiques. Le premier exemple est la synthèse des nitriles de Kolbe.
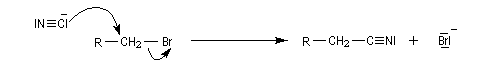

- alcoolates
- phénolates
- ammoniac et amines
- imides (synthèse de Gabriel des amines primaires)
- énamines (alkylation régiosélective de Stork)
- triphénylphosphine (préparation des ylures)
- dithianes lithiés (réaction de Corey-Seebach)
- phosphites d'alkyle (réaction d'Arbusov)
- ions alcynure
- ions carboxylate
- énolates de composés carbonylés
- énolates d'esters (synthèse malonique et synthèses apparentées)
- énolates d'amides
Des substitutions nucléophiles intramoléculaires peuvent intervenir avec rétention de configuration. Un exemple de tel mécanisme SNi s'observe lors de la réaction entre un alcool et le chlorure de thionyle dans l'éther en l'absence de pyridine.
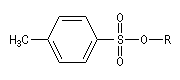
Dérivés sulfonylés
Notons que dans ce type de réaction, les dérivés halogénés sont avantageusement remplacés en tant que substrats par des mésylates, des tosylates ou des triflates obtenus par sulfonylation des alcools.
Les nucléofuges sont des bases beaucoup plus faibles donc plus facilement déplaçables que les ions halogénures. Les triflates d'alkyle sont, à cet égard, d'excellents substrats car l'ion triflate est la base conjuguée, très stable, de l'acide triflique, un superacide.
|
Mésylate (ROMs)
|
Triflate (ROTf)
|
Tosylate (ROTs)
|
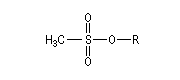 |
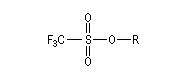 |
 |
Introduction
Pour un certain nombre de réactions de substitutions, on observe les résultats expérimentaux suivants :
- la vitesse de la réaction est indépendante de la concentration en nucléophile ;
- la réaction possède un ordre égal à 1 à l'instant initial ;
- l'ajout d'ion X- (nucléofuge) en cours de réaction, diminue la vitesse (effet de sel) ;
- réalisée à partir d'un substrat chiral pur, on obtient généralement un mélange racémique des deux énantiomères. On dit qu'il y a eu racémisation. De plus, des substrats énantiomères donnent généralement le même mélange racémique. La réaction n'est pas stéréospécifique. L'aspect stéréochimique est examiné dans un paragraphe particulier.
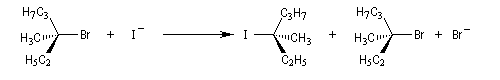
Le mécanisme suivant a été proposé :
- formation d'un du carbocation ;
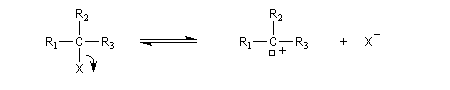
- réaction du carbocation avec le nucléophile.
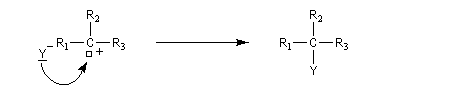
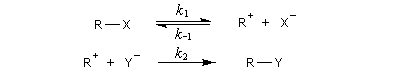
Par définition, la vitesse de la réaction est :
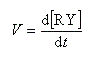
La deuxième étape est élémentaire donc l'ordre est égal à la molécularité de cette étape c'est à dire 2 (loi de Van 't Hoff.)
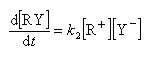
Exprimons la vitesse de formation du carbocation à partir des étapes élémentaires de formation et de destruction.
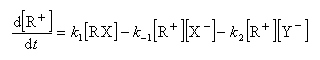
Cet intermédiaire de réaction ne s'accumule pas et on peut lui appliquer le principe de l'état quasi-stationnaire :

soit :
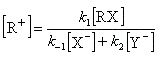
l'expression de la vitesse est donc :
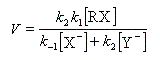
Cette expression montre qu'un ajout d'ions X- fait rétrograder la vitesse conformément à l'expérience (effet de sel). La réaction ne possède pas d'ordre.
Au début de la transformation, on peut négliger le premier terme du dénominateur devant le second.
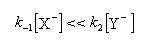
On observe alors expérimentalement un ordre égal à 1.
Le profil énergétique de la réaction est le suivant.
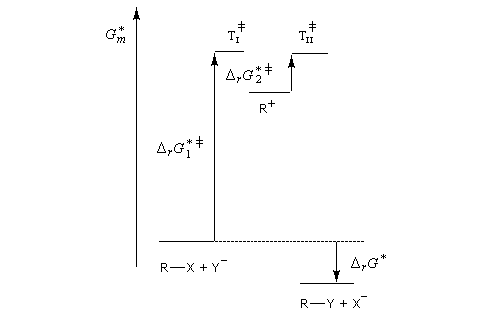
Puisqu'il se forme un intermédiaire de haute énergie, on peut appliquer le postulat de Hammond. Celui-ci nous indique que les deux états de transition ayant une énergie proche de celle du carbocation auront une structure voisine de celui-ci. En conséquence, tout facteur stabilisant le carbocation abaisse l'énergie de l'état de transition qui y conduit, donc accroit la vitesse de la réaction.
Effets stériques
Le passage de la géométrie tétraédrique de l'atome de carbone d'un dérivé halogéné à la géométrie plane de l'atome de carbone d'un carbocation s'accompagne d'une diminution des répulsions entre les liaisons car les angles entre-elles passsent de 109 ° à 120 °. Cette décompression stérique est maximale lorsque le dérivé halogéné est tertiaire.
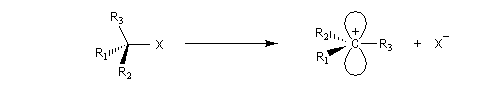
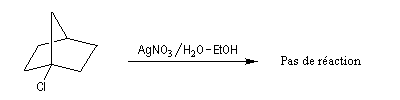
 |
La figure de gauche représente le 1-bromobicyclo[2,2,1]heptane ou 1-bromonorbornane. Lorsqu'on mélange ce composé avec une solution hydroalcoolique de nitrate d'argent, on n'observe pas de réaction même à chaud. Cela montre que ce composé n'est pas du tout réactif dans les réactions de substitutions nucléophiles SN1 car cela conduirait à un carbocation non plan. Notons que ce composé ne réagit pas non plus selon un processus SN2 à cause de l'encombrement stérique de l'atome de carbone porteur de l'halogène. |
On s'intéresse aux vitesses relatives de solvolyse dans l'acide méthanoïque pour la réaction suivante (100 °C).
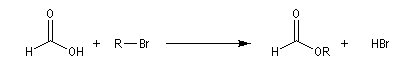
|
Composé
|
CH3Br
|
CH3CH2Br
|
(CH3)2CHBr
|
(CH3)3CBr
|
|
Vitesse relative
|
0,6
|
1,0
|
25
|
108
|
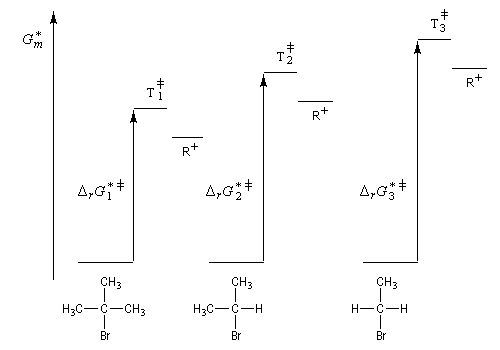
- l'effet inductif +I des groupes alkyles disperse la charge positive qui est de ce fait mieux supportée ;
- l'accroissement du nombre des substituants augmente les possibilités d'hyperconjugaison.
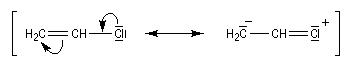
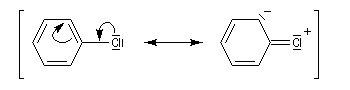
- addition-élimination ;
- élimination-addition avec formation d'un intermédiaire benzyne.
|
Composé
|
CH3CH2OTs
|
iPrOTs
|
CH2=CH-CH2OTs
|
PhCH2OTs
|
|
Vitesse relative
|
1
|
2,7
|
33
|
385
|
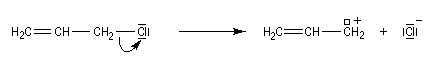
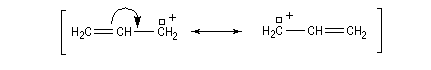
Les systèmes benzyliques se comportent de façon comparable. Les possibilités de délocalisation sont encore plus élevées.
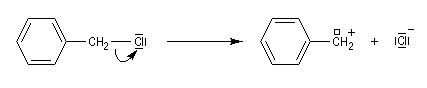
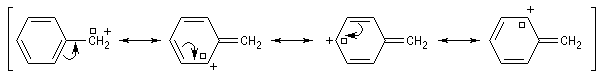
Afin d'étudier l'influence du solvant, il faut examiner comment il intervient dans l'étape d'ionisation du substrat qui est cinétiquement déterminante :
- un moment dipolaire élevé favorise l'ionisation ;
- une grande permittivité diélectrique augmente la dissociation des paires d'ions formées ;
- un solvant protique, donneur de liaison hydrogène, permet une bonne solvatation de l'anion.
|
Solvant
|
Permittivité relative er
|
Constante de vitesse relative k
|
|
CH3CO2H
|
6
|
1
|
|
CH3OH
|
33
|
4,0
|
|
HCO2H
|
58
|
5,0´103
|
|
H2O
|
78
|
1,5´105
|
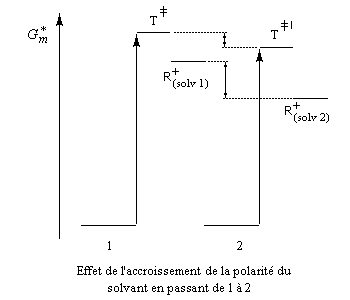
- une stabilisation de l'état de transition porteur de charges partielles ;
- une stabilisation plus élevée encore pour le carbocation et l'ion halogénure car ils sont porteurs de charges nettes. D'après le posutlat de Hammond, l'état de transition qui précède le carbocation voit également son énergie abaissée. Comme il s'agit de l'étape cinétiquement déterminante, la vitesse est accrue.
L'ajout d'acides de Lewis comme Ag+ ou AlCl3 qui forment des complexes acide-base de Lewis avec l'ion halogénure facilite l'ionisation du substrat. La vitesse de la réaction avec un nucléophile est accrue.
|
Classe du dérivé halogéné
|
I
|
II
|
III
|
|
Réaction
|
lente à chaud
|
lente à la température ambiante
|
instantanée à la température ambiante
|
La réaction "normale" entre un dérivé halogéné et un ion cyanure conduit à un nitrile par un mécanisme de type SN2. L'ion cyanure réagit par son pôle carboné mou, sur l'atome de carbone mou du substrat.
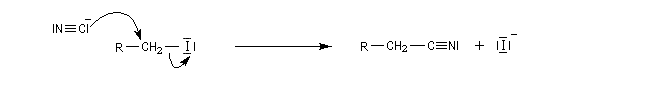
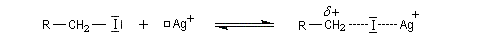
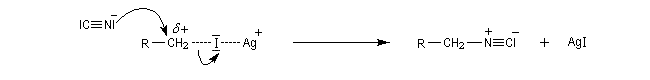
Dans l'introduction à l'étude des substitutions nucléophiles SN1, nous avons vu qu'en partant d'un substrat chiral, ces réactions conduisent généralement à un mélange racémique.
Un mécanisme plus fidèle à la réalité fait intervenir des paires d'ions. Dans ce qui suit, (s) désigne le solvant.
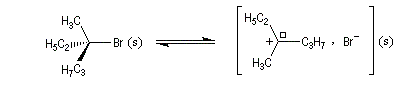
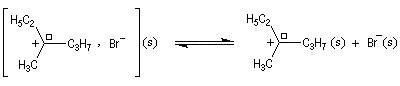
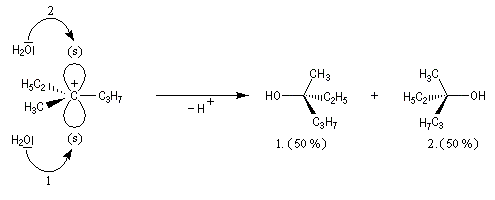
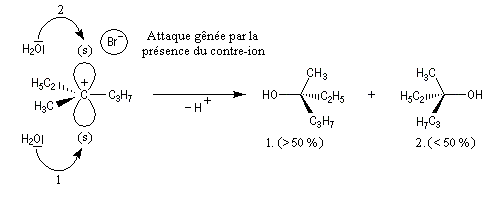
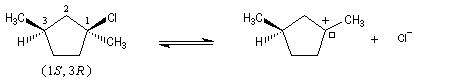
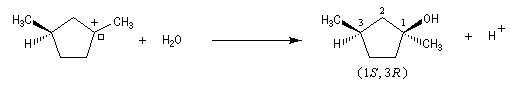

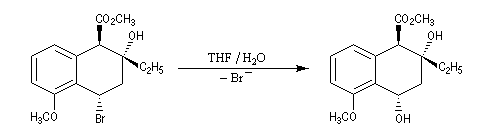
Transpositions de carbocations
Dans une réaction de transposition, un atome ou un groupe d'atomes se déplace d'un point à un autre. Lorsqu'il y a migration d'une liaison sigma, il s'agit d'une transposition sigmatropique.
Puisque les carbocations peuvent être générés facilement à partir d'alcools tertiaires, on rencontre là aussi ce type de transposition.
. Réactions d'éliminations
Dans une réaction d'élimination, deux groupes appartenant à une molécule et généralement portés par des atomes différents, sont éliminés (les a-éliminations constituent un cas particulier). Il y a formation d'une insaturation. Celle-ci peut être une liaison double ou correspondre à un cycle.
Eliminations b bimoléculaires E2
Introduction
Les réactions d'élimination b initiées par des bases à partir des dérivés halogénés produisent des alcènes. On peut les regarder comme les réactions inverses des additions électrophiles des halogénures d'hydrogène sur la double liaison éthylénique.
Stéréosélectivité, stéréospécificité
La réaction d'élimination nucléophile bimoléculaire est diastéréospécifique. Afin que les orbitales à l'origine de la double liaison puissent se recouvrir efficacement, les liaisons impliquées dans le processus d'élimination doivent être dans un même plan. Lorsque les contraintes au sein de la molécule le permettent, la disposition favorable des liaisons correspond à une géométrie antipériplanaire.
-
le composé de configuration absolue (2R,3S) fournit le composé éthylénique de configuration relative Z ;
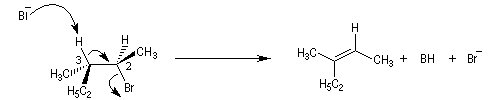
- le composé de configuration absolue (2R,3R) fournit le composé éthylénique de configuration relative E.
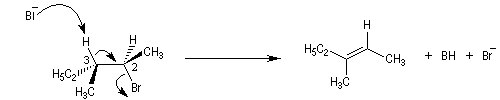
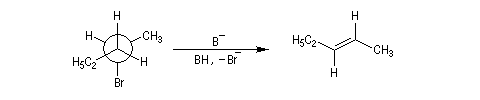
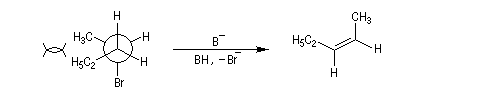
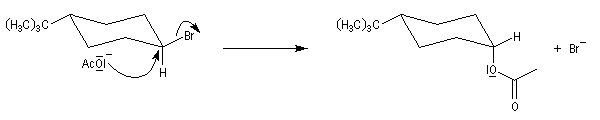
En série cyclohexanique, le fait que les liaisons doivent adopter une position antipériplanaire implique que les liaisons soient diaxiales. Une molécule rigide comme un dérivé bromé du tertiobutylcyclohexane en a du groupe tertiobutyle, ne conduit à une élimination facile qu'avec le dérivé cis.
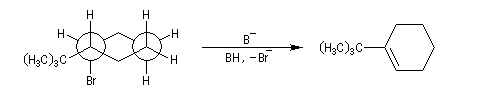
Un autre exemple concerne l'élimination sur le chlorure de menthyle (I) et le chlorure de néomenthyle (II).
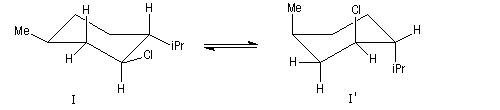
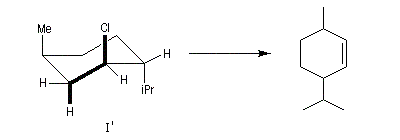
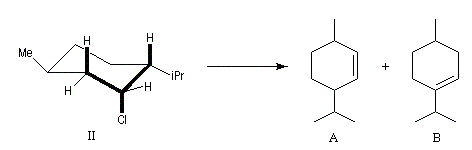
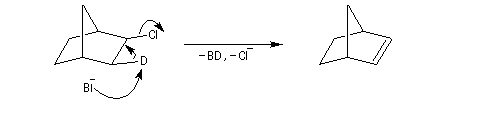
- élimination à partir des oxydes d'amines (réaction de Cope) ;
- pyrolyse des xanthogénates (réaction de Tschugaeff) ;
- élimination à partir des oxaphosphétanes dans la réaction de Wittig ;
- élimination à partir des b-hydroxysilanes en milieu basique (élimination de Peterson).
La transformation étant irréversible, les déshydrohalogénations E2 sont sous contrôle cinétique. Le produit majoritaire est le plus vite formé. On observe que le composé éthylénique le plus substitué est majoritaire car la double liaison est déjà partiellement formée dans l'état de transition (règle empirique de Zaytzev).
- le pourcentage d'alcène le plus substitué augmente lorsque le caractère nucléofuge augmente (tendance E1) ;
- il diminue avec l'augmentation de l'encombrement de la base (tendance E1cB).
Influence de la nature de la base
Pour promouvoir les éliminations E2 on utilise des bases fortes (HO-, RO-, H2N- ). On a aussi recours à des amines encombrées non nucléophiles (DBU, DBN, DABCO).
Influence du solvant
Il ne faut pas utiliser de solvant trop protique de façon à laisser à la base sa pleine capacité d'action. C'est la raison pour laquelle on utilise de préférence KOH dissous dans l'éthanol plutôt que dans l'eau. Certaines composés ioniques comme tBuOK voient leur activité basique renforcée par l'utilisation d'agents de transfert de phase comme les éthers couronnes ou les cryptands. La complexation de l'ion K+ exalte la basicité de l'anion.
Compétition entre les mécanismes E2 et SN2
L'élimination E2 et la substitution SN2 sont toujours en compétition l'une avec l'autre. Examinons quelques résultats expérimentaux.
- alkylation de l'ion éthanoate (acétate) par divers dérivés halogénés (solvant : propanone)
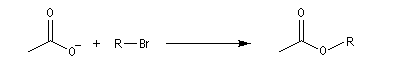 Dérivé halogénéPourcentage de substitutionPourcentage d'éliminationCH3CH2Br1000(CH3)2CHBr1000(CH3)3CBr0100(CH3)2CHCHBrCH31189
Dérivé halogénéPourcentage de substitutionPourcentage d'éliminationCH3CH2Br1000(CH3)2CHBr1000(CH3)3CBr0100(CH3)2CHCHBrCH31189 - alkylation de l'ion éthanolate, base plus forte que la précédente, (synthèse de Williamson) par divers dérivés halogénés (solvant : éthanol)
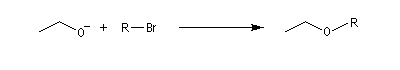
|
Dérivé halogéné
|
Pourcentage de substitution
|
Pourcentage d'élimination
|
|
CH3CH2Br
|
99
|
1
|
|
(CH3)2CHBr
|
20
|
80
|
|
(CH3)2CHCH2Br
|
40
|
60
|
|
CH3CH2 CHBrCH2CH3
|
12
|
88
|
- Les halogénures primaires donnent de façon majoritaire des réactions de substitution par un mécanisme SN2. On peut promouvoir l'élimination E2 en utilisant des bases fortes non nucléophiles.
- Les halogénures secondaires se comportent comme les primaires mais en présence d'un très bon nucléofuge, on observe des mécanismes SN1 et E1 avec des bases faibles.
- Les dérivés tertiaires ne donnent pas de substitution SN2. Ils donnent des réactions SN1 avec des bases faibles en compétition avec l'éliminations E1. Avec des bases fortes, on observe des éliminations E2.
Expérimentalement, on constate qu'une élévation de la température favorise l'élimination aux dépens de la substitution. On peut rationaliser ce résultat en remarquant que dans une réaction de substitution une liaison est rompue et une autre est reformée tandis que dans une réaction d'élimination, deux liaisons sont rompues et une liaison double est formée. L'énergie d'activation d'une réaction d'élimination est donc plus élevée que celle d'une réaction de substitution. L'élimination ne devient donc prédominante par rapport à la substitution que si la température est suffisamment élevée.
Applications
Lorsqu'on réalise une réaction d'élimination sur un dérivé dihalogéné, on obtient dans un premier temps un halogénure vinylique. Si l'on poursuit la réaction, on peut obtenir un diène 1,3. L'exemple suivant est une étape de la synthèse de Schenk de la cantharidine.
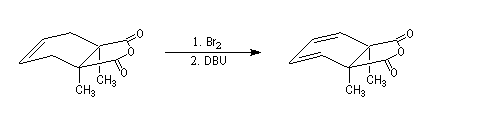
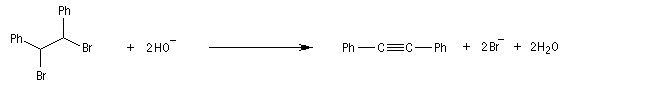
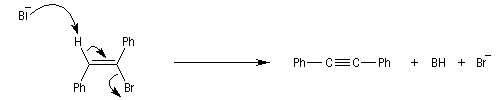
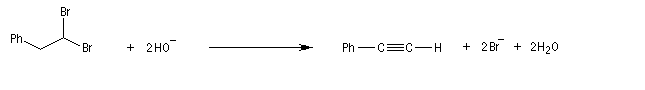
Introduction
On effectue la solvolyse du 2-bromo-2-méthylpropane (bromure de tertiobutyle) dans un mélange éthanol-eau. A côté du produit de substitution normalement attendu qui est l'alcool, on observe la formation d'un composé éthylénique. Ce dernier produit est le fruit d'une réaction d'élimination.
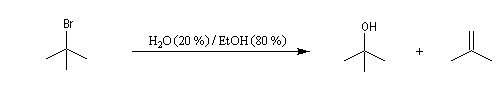
Le mécanisme suivant en deux stades permet de rendre compte des résultats expérimentaux :
- l'ionisation du substrat avec formation d'un carbocation est l'étape cinétiquement déterminante ;
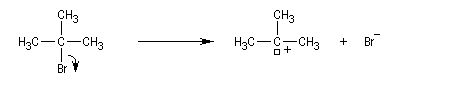
- un proton est ensuite arraché du carbocation dans une seconde étape rapide.
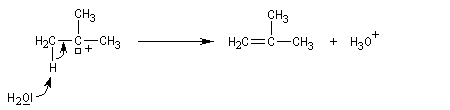
Régiosélectivité
On s'intéresse à la solvolyse du 2-bromo-2-méthylbutane dans un mélange éthanol eau 80/20.
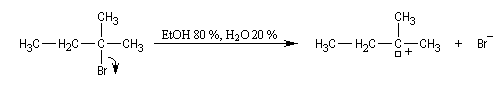
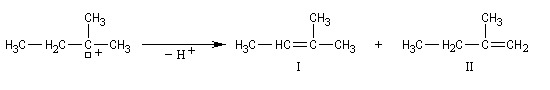
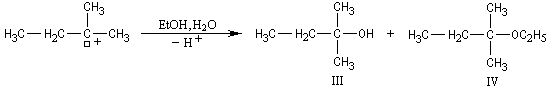
|
Dérivé halogéné
|
I
|
II
|
III + IV
|
|
Pourcentage
|
32
|
8
|
60
|
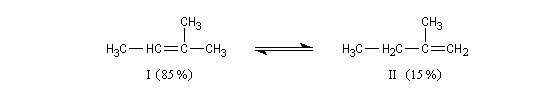
Compétition entre les mécanismes E1 et SN1
Puisque la première étape de leur mécanisme respectif est commune, l'élimination E1 est toujours en compétition avec la substitution SN1. Les facteurs qui influencent l'un ou l'autre des deux mécanismes sont assez semblables à ceux qui interviennent pour déterminer le rapport SN2/E2. L'élimination est favorisée par :
- une élévation de température ;
- une augmentation de la force de la base.
Eliminations b E1cB
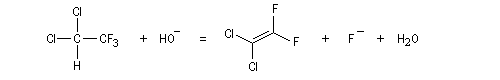
Introduction
Lorsqu'on fait réagir le 1,1-dichloro-2,2,2-trifluoroéthane avec une solution aqueuse d'hydroxyde de sodium, on observe la réaction suivante.
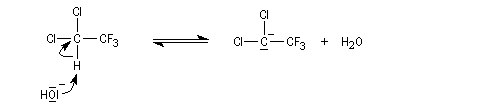
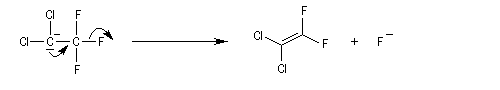
Théorie de l'état de transition variable
Les résultats expérimentaux montrent que l'élimination E2 fait intervenir un ensemble de mécanismes qui diffèrent les uns des autres par la synchronisation plus ou moins élevée des ruptures des liaisons C-H et C-X. Dans la théorie de l'état de transition variable développée par le chimiste américain J. F. Bunnett, le mécanisme E2 synchrone occupe le centre du spectre tandis que les limites correspondent aux mécanismes E1Cb et E1 respectivement [13].
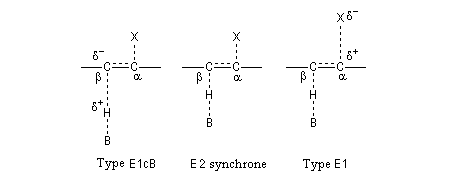
- Dans le type E1cB (E1cB like), la rupture de la liaison C-H précède celle de la liaison C-X. A la limite, on a la formation d'un carbanion ;
- Dans le mécanisme E2, les liaisons sont rompues de façon synchronisée ;
- Dans le type E1 (E1 like), la rupture de la liaison C-X précède celle de la liaison C-H. A la limite, on a la formation d'un carbocation.
- la nature de la base ;
- la nature du nucléofuge ;
- la structure du substrat et notamment l'influence électronique des substituants ;
- la nature du solvant.
a-élimination
Dans une réaction d'a-élimination, les deux groupes éliminés sont liés au même atome de carbone. Il y a formation d'un carbène (ou d'un intermédiaire analogue). L'existence des carbènes comme intermédiaires de réaction a été postulée par J. Hine en 1950 lors de son étude de la réaction entre le chloroforme et les ions hydroxyde [15].
Le passage des ions hydroxyde en phase organique est facilité par l'utilisation d'un catalyseur par transfert de phase.
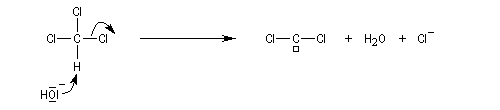
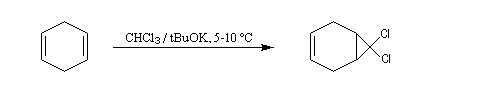
La réaction de Reimer-Tiemann constitue un autre exemple d'utilisation de dihalogénocarbènes en synthèse.
Notons que des dérivés organométalliques de carbènes interviennent comme intermédiaires dans la métathèse des composés éthyléniques.
g-élimination
Un exemple est la déshalogénation par un métal comme le zinc des dérivés 1,3-dihalogénés conduisant au cyclopropane.
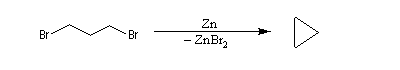
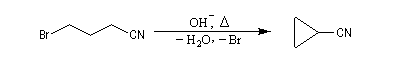
Des électrons de liaisons multiples peuvent assurer un relais dans le déplacement des liaisons. La réaction suivante est une méthode de synthèse de diènes conjugués. Une autre voie d'accès à ces composés consiste à effectuer deux éliminations b successives.
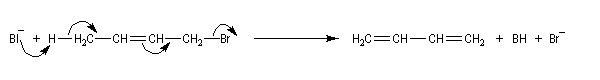
Il s'agit d'une réaction d'élimination des ions carboxylate des acides b-halogénés. Elle est utilisée dans la synthèse stéréospécifique des composés éthyléniques.
Elimination utilisant des métaux
Les dérivés dihalogénés traités par un métal comme le zinc ou le magnésium conduisent au composé éthylénique par une réaction d'élimination. Il y a formation d'un composé organométallique intermédiaire. L'exemple suivant concerne la synthèse du cyclobutène [34].
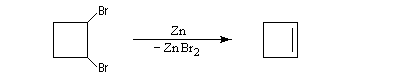
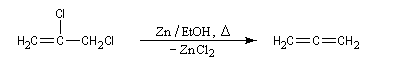
A partir du 1,3-dibromopropane, on obtient le cyclopropane par une réaction de g-élimination.
Couplages utilisant des métaux de transition comme catalyseurs
Introduction
L'importance des réactions de couplage catalytique des dérivés halogénés utilisant des complexes de métaux de transition comme ceux du palladium, a été récemment illustrée par l'attribution du prix Nobel de chimie 2010 au chimiste américain R. Heck et aux chimistes japonais Negishi et Suzuki. Historiquement, la première réaction de couplage croisé à avoir été publiée en 1972 est la réaction de Kumada entre un dérivé halogéné et un organomagnésien.
Réaction de Mozoroki-Heck
Cette réaction a été découverte indépendamment par le chimiste japonais T. Mizoroki et l'américain R. Heck dans les années 1970 [16].
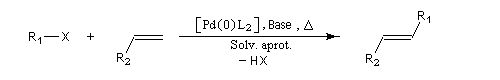
X : halogène (I, Br, Cl) , OTf (triflate) ;
R2 : alkyle, alcényle, aryle, allyle, CO2R ; OR, SiR3.
Le composé éthylénique doit posséder au moins un atome d'hydrogène vinylique.
Le catalyseur est un complexe du palladium (0) comme le tris(dibenzylidèneacétone)dipalladium (0) Pd2(dba)3 qui est une source stable de Pd (0). On peut aussi utiliser un composé du Pd (II) tel que Pd(OAc)2 en présence d'une phosphine telle que PPh3 permettant de générer Pd (0) in situ.
Un solvant aprotique tel que (THF, DMF) (mais des procédures en milieu aqueux existent)
La base est typiquement une amine tertiaire telle que Et3N.
La température est comprise dans la gamme : 20 °C à 140 °C.
La réaction est stéréosélective. On obtient de façon préférentielle le composé éthylénique de stéréochimie E. L'aspect mécanistique de la réaction de Heck est différent de celui des réactions de Suzuki et de Negishi. Il existe en fait deux mécanismes possibles :
- mécanisme neutre ;
- mécanisme cationique.
Réaction de Negishi
Cette réaction été mise au point par le chimiste japonais Negishi en 1977 [50]. Celui-ci a obtenu le prix Nobel de chimie en 2010 pour cette découverte [36].
C'est une réaction de couplage croisé catalysée par un complexe du palladium, entre un dérivé halogéné R1X et un organozincique R2ZnX'.
R1 : alcényle, aryle, allyle, alcynyle.
X : halogène (I, Br, Cl) , OTf (triflate) ;
R2 : alkyle, alcényle, aryle, allyle ;
X : halogène (I, Br, Cl)
Un complexe du palladium (0) tel que Pd(PPh3)4 est utilisé en quantité catalytique.
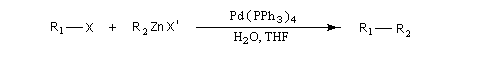
La présence du zinc permet à la réaction d'être compatible avec de nombreux groupes fonctionnels (alcool , phénol, silanes, acétates, ester boroniques, amides).
Le mécanisme simplifié est résumé ci-dessous.
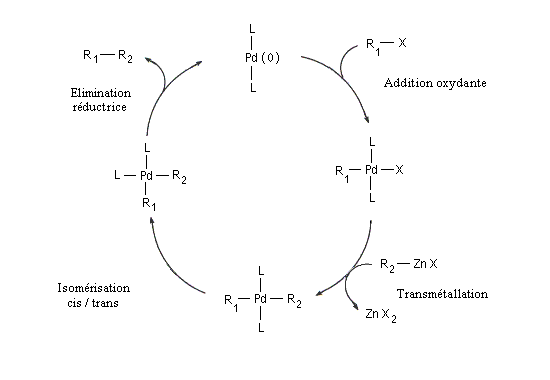
La réaction de Negishi a notamment été utilisée avec succès pour synthétiser des biaryles non symétriques encombrés qui sont traditionnellement difficiles d'accès.
Naguère, la synthèse des biaryles symétriques était réalisée presque exclusivement grâce au couplage d’Ullmann. La réaction suivante est une variante énantiosélective utilisant comme catalyseur, le tris(dibenzylidèneacétone)dipalladium (0) et comme catalyseur chiral un dérivé du ferrocène. Elle permet la préparation d'un binaphtyle chiral avec une très bonne énantiosélectivité. (P. Espinet et coll.)
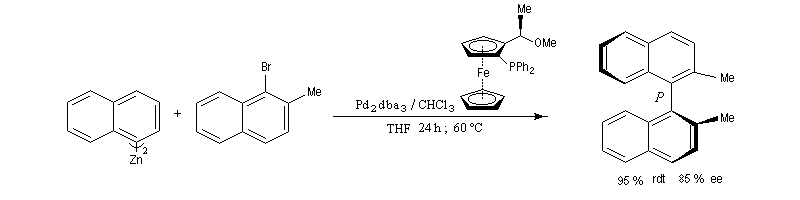
Réaction de Suzuki
Cette réaction a été proposée par N. Miyaura et A. Suzuki en 1979 [51]. Ce dernier a obtenu le prix Nobel de chimie en 2010 pour cette découverte [36].
Il s'agit d'une réaction de couplage croisé entre un électrophile R1X : dérivé halogéné vinylique ou arylique ou encore un dérivé sulfonylé (triflate) et un acide boronique R2B(OH)2 ou mieux, un ester boronique R2B(OR')2 ou encore un trialkylborane avec :
R1 : alcényle, aryle, benzyle, allyle, alcynyle ;
X = halogène (I, Br), OTf (triflate), N2+ ;
R2 : phényle, alcényle.
Un complexe du palladium tel que le Tétrakis (triphénylphosphine)palladium (0) : Pd(PPh3)4, est utilisé en quantité catalytique.
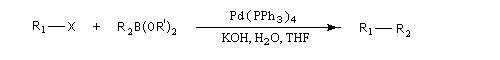
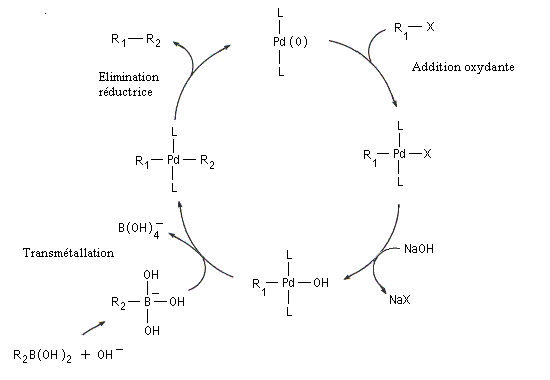
L’activation par une base de l’entité boronique facilite l'étape de transmétallation. L'organoborane est habituellement préparé par hydroboration d'un alcyne ou par hydroboration d'un alcène.
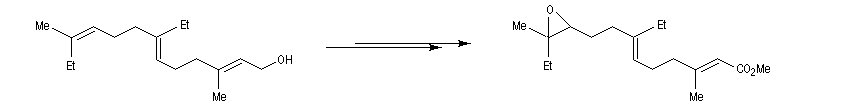
Un très gros avantage de ce type de réaction est que les réactifs peuvent posséder des groupes fonctionnels variés. Il est ainsi possible de coupler entre-elles des chaînes carbonées fonctionnalisées de grandes tailles lors d'une synthèse. Dans l'exemple suivant, l'une des chaînes de l'organoborane comporte un époxyde pourtant réputé comme étant particulièrement réactif.
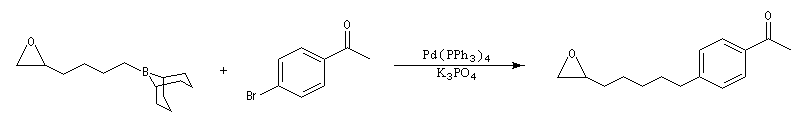
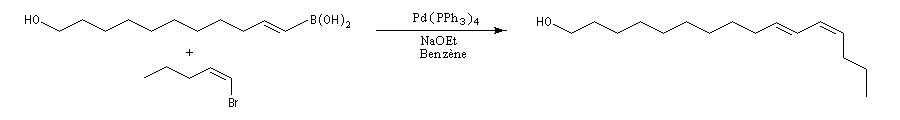
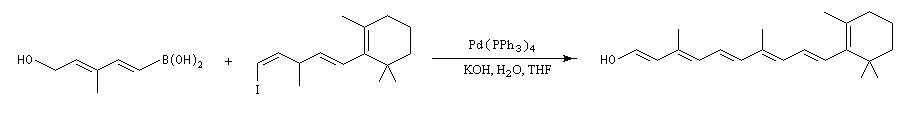
La dernière étape de la synthèse de la myxalamide A par C.H. Heathcock est une réaction de couplage de Suzuki entre un vinylborane de stéréochimie E et un iodotriene fonctionnalisé de stéréochimie Z [52]
Le coût assez modique des réactifs de départ permet une utilisation à l'échelle industrielle.
Alkylations de composés organométalliques
Réaction de Kumada
Cette réaction d'alkyation des organomagnésiens par un dérivé halogéné en présence d'un complexe de nickel ou de palladium est étudiée dans le chapitre relatif aux composés organométalliques.
Réaction de Corey-House-Posner-Whitesides
L'alkylation des cuprates lithiens par des dérivés halogénés primaires est connue sous le nom de réaction de Corey-House-Posner-Whitesides. Elle permet la fixation de groupes alkyles à partir de substrats comportant des halogènes comme l'iode qui est un bon groupe nucléofuge [49].
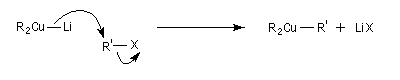
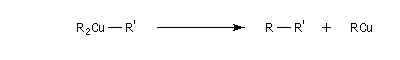
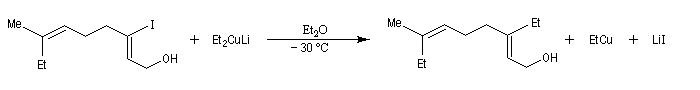
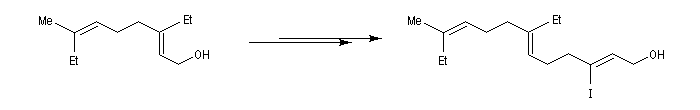
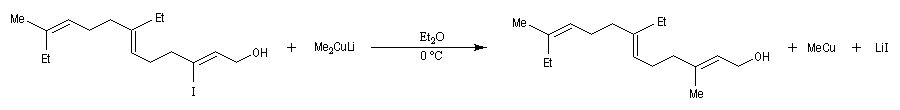
Alkylation des alcénylcuprates
L'alkylation directe des organomagnésiens vinyliques n'est pas possible sans catalyse en raison du manque de réactivité de la liaison métal-halogène. Les alcénylcuprates ou organocuprates vinyliques, sont susceptibles d'être directement alkylés avec un iodure d'alkyle. Ce type de réaction constitue une méthode de synthèse d'alcènes substitués.
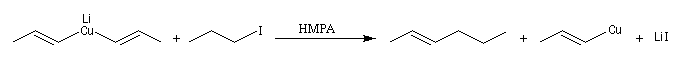
Les cuprates de Lipshutz permettent l'alkylation de dérivés halogénés secondaires.
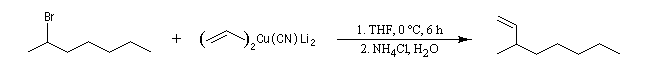
La réaction repose sur la formation d'un organosodique.
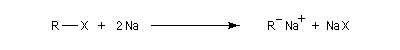
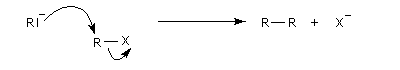
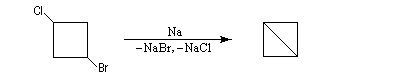
Réactions de réduction
Réduction par les métaux
La réaction entre un dérivé halogéné et le zinc conduit à un organozincique. Ce dernier est ensuite traité par un donneur de proton comme l'acide acétique.
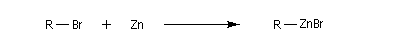
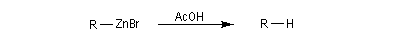
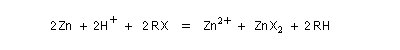
Réduction par les hydrures
LiAlH4 permet la réduction des dérivés halogénés. La réaction s'apparente à une substitution nucléophile par l'ion hydrure. On utilise aussi LiBHEt2 qui s'avère très efficace dans cette réaction.
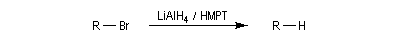
Les dérivés halogénés sont réduits par le dihydrogène en milieu basique.
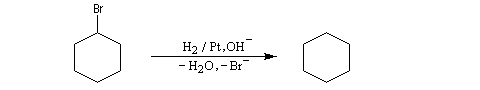
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D.C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C.A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B.C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
Articles
[13] J. F. Bunnett, Angew. Chem. Int. Ed. Engl. 1, 225 (1962).
[14] Sir Christopher Ingold- A Major Prophet of Organic Chemistry by Kenneth T. Leffek, JCE, June 1997 Vol. 74 No. 6 p. 625
[15] Hine J, J. Am. Chem. Soc. 1950, 72, 2438.
[16] Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides, R. F. Heck, J. P. Nolley, J. Org. Chem., 1972, 37 (14), pp 2320–2322
[49] Posner, G. H. Substitution Reactions using Organo Copper Reagents, Organic Reactions 1975, 22, 253.
[50] Anthony O. King, Nobuhisa Okukado and Ei-ichi Negishi J. Chem. Soc., Chem. Commun., 1977, 683-684
[51] Miyaura, N.; Suzuki, A. Chem. Commun. 1979, 866.
[52] Mapp, A.K., Heathcock, C. H. Total Synthesis Myxalamide A.J. Org. chem. 1999, 64,23-27.
Liens
[17] Nucleophilic substitution
[18] Chemistry Comes Alive, nucleophilic substitution
[19] The Hard Soft Acide Base Principle
[20] Tyrian purple
[21] Cours sur les dérivés halogénés
[22] n-hexadecane by P. A. Levene
[23] linoleic acid by J. W. McCutcheon
[25] Benzocyclopropene by W. E. Billups, A. J. Blakeney, and W. Y. Chow
[26] 1,6-methano-10-annulene by E. Vogel, W. Klug, and A. Breuer
[27] Bicyclo[1,1,0]butane by Gary M. Lampman and James C. Aumiller
[28] Rearrangements induced by cationic or electron deficient sites by William Reusch
[29] Cyclopropanecarboxylic acid by Chester M. McCloskey and George H. Coleman
[30] Glossary of Terms used in Physical Organic Chemistry IUPAC Recommendations 1994
[31] Diphenylacetylene by Lee Irvin Smith and M. M. Falkof
[32] 1,1-dichloro-2,2-difluoroéthylène by J. C. Sauer
[33] Allène by H. N. Cripps and E. F. Kiefer
[34] Cyclobutene by J. Salaün and A. Fadel.
[35] (Z)-1-iodohexene by A. Alexakis, G. Cahiez, and J. F. Normant.
[36] Prix Nobel de chimie 2010
[37] Thèse P. E. Broutin.
[38] Thèse L. Chahen.
[39] Synthesis by metal mediated Coupling Reactions
[40] Heck reaction: Mechanism and it’s application in total synthesis of Natural Products by Venugopal Rao
Ouvrages théoriques
A. Streitwieser Jr, C.H. Heathcock - Introduction to Organic Chemistry (Macmillan Publishing Co).
J. March - Advanced organic chemistry (Wiley Interscience 2006).
F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, (Plenum Press, 1990).
N. T. Ahn - Introduction à la chimie moléculaire, (Ellipses, 1994).
C. Reichardt - Effets de solvants en chimie organique, (Flammarion Sciences, 1971).
P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique (Ellipses, 1993).
P. Atkins - General Chemistry (Scientific American Books W.H Freeman and Co).
Metal-catalyzed Cross-coupling Reactions, Edited by F. Diederich and P. J. Stang, Wiley-VCH, New York, 2004
Organometallics in Synthesis. A manual, Schlosser, M., Second Edition, Wiley, New York, 2002.
Pour aller plus loin
Suzuki cross coupling
Synthèse organique par voie organométallique par T. Ollevier
Organometallic compound of Lithium Sodium and Potassium by Michael K. Denk.
Organometallic chemistry by Worawan Bhanthumnavin
Bien qu'étant assez anciens, les ouvrages suivants conservent de l'intérêt :
C. K. Ingold - Structure and Mechanism in Organic Chemistry, Cornell Univ. Press, Ithaca (1953).
J. Hine, Physical Organic Chemistry, 2nd Edition, McGraw-Hill, New York (1962).
R. Breslow - Mécanismes des réactions organiques (Edisciences, - 1970).










0 commentaires:
Enregistrer un commentaire