Cours de chimie Organique -
Ethers acycliques et époxydes
Nomenclature
Ethers non cycliques
On appelle éther un composé dans lequel un atome d'oxygène est lié par liaison simples à deux groupes organiques différents. La nomenclature recommandée par l'UICPA est la nomenclature substitutive. Les éthers simples sont nommés comme des alkoxyalcanes. La nomenclature radico-fonctionnelle est encore largement utilisée surtout pour les éthers les plus simples. A ces noms systématiques, il convient d'ajouter les noms usuels. Ainsi l'éthoxyéthane est appelé couramment éther ordinaire ou éther sulfurique par référence à son mode d'obtention.
|
I
|
II
|
III
|
IV
|
|
méthoxyméthane
|
méthoxyéthane
|
éthoxyéthane
|
2-éthoxypropane
|
|
oxyde de diméthyle
|
oxyde de méthyle et d'éthyle
|
oxyde de diéthyle
|
oxyde d'éthyle et de 2-méthyléthyle
|
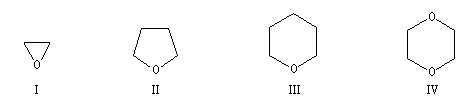
Le nom de base est celui du cyclane. Les atomes d'oxygène qui remplacent ceux de carbone dans le cycle sont indiqués par oxa.
|
I
|
II
|
III
|
IV
|
|
oxacyclopropane
|
oxacyclopentane
|
oxacyclohexane
|
1,4-dioxacyclohexane
|
|
oxyde d'éthène
|
tétrahydrofurane (THF)
|
tétrahydropyrane (THP)
|
dioxane
|
Propriétés physiques
Caractéristiques géométriques et énergétiques
L'atome d'oxygène des éthers contracte deux liaisons simples avec les atomes de carbone des groupes organiques liés. Il reste donc deux paires d'électrons non liantes sur l'atome d'oxygène. La géométrie localement plane autour de l'atome d'oxygène dérive d'un arrangement tétraédrique des paires d'électrons. Puisque les paires non liantes occupent en moyenne un volume plus grand que les paires liantes, on prévoit un angle entre les liaisons supérieur à celui observé dans un tétraèdre régulier c'est à dire a >109°. Le tableau suivant regroupe quelques valeurs moyennes de grandeurs géométriques et énergétiques. Remarquons la valeur élevée de l'énergie de dissociation de la liaison C-O.
|
d (C-O) (nm)
|
a (COC) (°)
|
Do (C-O) (kJ.mol-1)
|
|
0,141
|
118
|
343
|
|
composé
|
constante diélectrique er
|
moment dipolaire m (D)
|
| éthoxyéthane |
4,2
|
1,25
|
| tétrahydrofurane |
7,4
|
1,70
|
|
nom
|
TF (°C)
|
TE (°C)
|
| méthoxyméthane |
-138,5
|
-23
|
| éthoxyéthane |
-116,62
|
34,5
|
Spectroscopie infrarouge
On distingue les éthers non cycliques, les éthers cycliques et dans cette famille les époxydes. Les valeurs suivantes correspondent aux éthers non cycliques.
|
vibration
|
C-O-C (asym)
|
C-O-C (sym)
|
|
nombre d'onde (cm-1)
|
1260-1070 (forte)
|
1055-870 (forte)
|
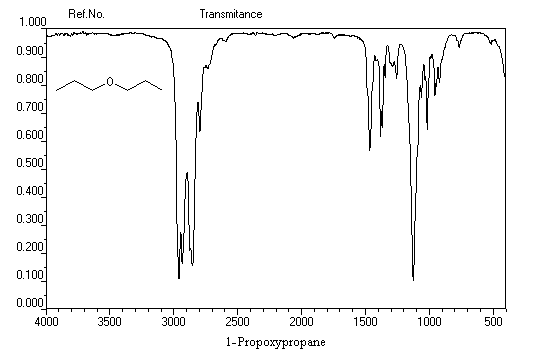
Ethoxyéthane
 |
L'éthoxyéthane est l'éther le plus utilisé comme solvant. Il est peu toxique (VME 1200 mg.m-3 ). Il est très volatil (TE = 34,6 °C) et extrêmement inflammable comme en témoigne un point éclair très bas (TE = -40°C). Il a été utilisé comme anesthésique. On peut l'obtenir par déshydratation intermoléculaire de l'éthanol en présence d'acide sulfurique à 130-140 °C.
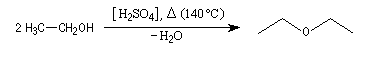 |
 |
Le 2-méthoxy-2-méthylpropane ou méthyltertiobutyléther
(MTBE) améliore l'indice d'octane des supercarburants qui peuvent en
contenir jusqu'à 13,6 % en masse. On l'obtient par réaction entre le
méthylpropène et le méthanol, catalysée par une résine acide (polystyrène sulfonique réticulé).
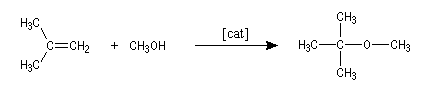 |
 |
Le tétrahydrofurane est un éther très utilisé comme
solvant en synthèse. Il a une température d'ébullition plus grande que
l'éthoxyéthane (TE = 67 °C). Il est narcotique et toxique (VME 590 mg.m-3).
On peut l'obtenir par déshydratation du butan-1,4-diol. Il est
difficile de le conserver sec car il est assez miscible avec l'eau.
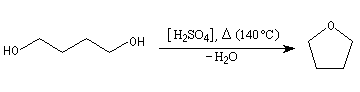 |
 |
Le dioxane est utilisé comme solvant. Il possède une
température d'ébullition assez élevée (TF = 11 °C, TE = 101,3 °C). C'est
un composé toxique. On peut l'obtenir par déshydratation intermoléculaire de l'éthylèneglycol sous l'action de l'acide sulfurique.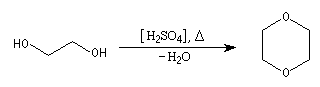 |
 |
On prépare ce composé par réaction entre l'éthène et le dioxygène à 170 °C en présence d'Ag comme catalyseur. A la température ordinaire c'est un liquide toxique (TE = 12,5 °C).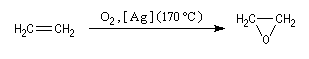 |
Séchage des éthers
Les éthers peuvent contracter des liaisons H avec l'eau ou les alcools avec lesquels ils sont miscibles dans une certaine proportion. L'utilisation de réactifs incompatibles avec l'eau même à l'état de traces comme les organométalliques pose le problème du séchage des éthers.
A la température ordinaire, le méthoxyméthane est miscible à l'eau en toutes proportions. L'éthoxyéthane, qui possède une chaîne carbonée plus longue, l'est nettement moins. Cependant il peut contenir jusqu'à 1,5 % d'eau.
Le THF est beaucoup plus miscible à l'eau que l'éther ordinaire. On l'attribue au fait que les doublets non liants de l'oxygène sont beaucoup plus accessibles pour contracter des liaisons H. Son séchage revêt une importance particulière en synthèse organique.
On distingue plusieurs étapes selon la quantité d'eau présente dans l'éther :
- Le séchage ordinaire peut être réalisé en utilisant des agents desséchants chimiques parmi lesquels : MgSO4 anhydre, Na2SO4 anhydre etc ou en faisant passer lentement l'éther dans une colonne remplie de tamis moléculaires.

Les zéolithes naturelles ont été découvertes par le minéralogiste A. F. Cronsted en 1756 qui leur a donné leur nom (pierre qui bouillent) car elles bouillonnent quand on les chauffe. Ces aluminosilicates possèdent une structure caractérisée par la présence de cages reliées entre-elles par des tunnels. On sait produire artificiellement des zéolithes possédant des cages de tailles calibrées. Elles peuvent servir à séparer les constituants d'un mélange par piégeage de molécules comme l'eau dans les cavités, d'où leur nom de tamis moléculaire. La photographie de gauche représente des tamis moléculaires dont les cavités font 0,4 nm (tamis 4 A) en extrudés de 1,6 mm. Ils sont utilisés pour le séchage des liquides organiques. On peut conserver les éthers sur ce type de tamis placé au fond de la bouteille. - Le THF anhydre peut être obtenu en agitant du THF préalablement séché par les méthodes précédentes avec des petits morceaux de sodium.
Attention : ne pas utiliser le sodium en présence d'une trop grande quantité d'eau en raison du risque de réaction violente entre le sodium et l'eau.
Lorsqu'on ajoute de la benzophénone et du sodium dans le THF rigoureusement anhydre, on observe la formation d'un anion-radical cétyle qui donne à la solution une coloration bleue intense. Le mécanisme de cette réaction est détaillé dans la partie consacrée à la réduction par les métaux des composés carbonylés.

Dans le ballon, surmonté d'un réfrigérant ascendant, on a placé du THF préalablement séché sur tamis moléculaire, des petits morceaux de sodium (coupés en dés de 5 mm environ) et une spatule de benzophénone. Le mélange est agité plusieurs dizaines de minutes sous la hotte. 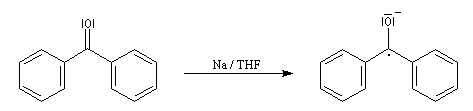
Cette manière de procéder combine l'avantage d'un séchage total et de l'élimination des peroxydes car ces derniers sont réduits par le sodium.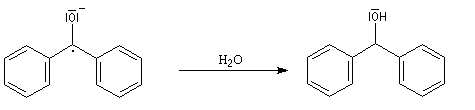
Extraction par solvant
L'éthoxyéthane est souvent utilisé comme solvant d'extraction car il dissout de nombreuses matières organiques et il est facile à éliminer. Un appareil simple utilisé pour les extractions liquide-liquide est l'ampoule à décanter.
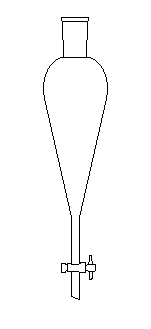 |
L'ampoule à décanter (qu'il ne
faut pas confondre avec une ampoule de coulée) est utilisée pour
l'extraction liquide-liquide. La solution S contenant le composé à
extraire et le solvant d'extraction S
(par exemple l'éther) sont introduits dans l'ampoule. Le composé à
extraire A se partage entre les deux solvants selon un équilibre qu'on
peut schématiser par l'équation :
A(S) = A(S )
Le rapport :
K = [A(S )]/[A(S)]
s'appelle coefficient de partage. A une température donnée, c'est une constante. L'extraction est d'autant plus complète que K est plus grand. Pour extraire la quantité maximale de composé d'une solution on a intérêt à procéder en plusieurs étapes. On réalise des extractions successives avec plusieurs fractions de solvant. Les phases organiques sont ensuite combinées. |
 |
 |
Photographie I : le mélange de la solution aqueuse de b -carotène et d'éther est agité vigoureusement. L'opérateur ouvre le robinet après chaque agitation pour équilibrer la pression interne et la pression atmosphérique. C'est l'opération d'extraction proprement dite. Photographie II : la phase organique constituée de b -carotène dans l'éther constitue la phase supérieure. La phase aqueuse qui ne contient presque plus de carotène constitue la phase inférieure. Ces phases se sont distribuées dans cet ordre par décantation. Elles peuvent ensuite être séparées avec précision grâce à la forme particulière de la partie terminale de l'ampoule. |
|
I
|
II
|
Elimination du solvant
Un des avantages de l'éther comme solvant d'extraction est qu'il peut être éliminé rapidement du fait de sa grande volatilité.
 |
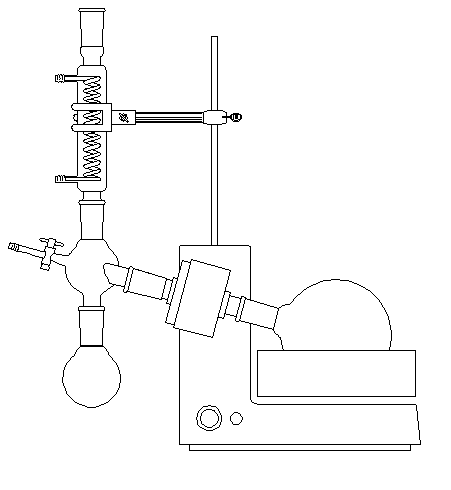
Un appareil couramment utilisé pour éliminer l'éther (ou plus généralement le solvant) d'un mélange, est l'évaporateur rotatif (appelé souvent "rotavapor"). Le mélange de solvant et de soluté est placé dans le ballon de droite. Celui-ci est plongé dans un bain-marie. Il est incliné et animé d'un mouvement de rotation de manière à créer un film de liquide et ainsi accroître la surface d'évaporation du solvant. La pression à l'intérieur du montage est abaissée au moyen d'une trompe à eau ce qui augmente la vitesse d'évaporation. Après condensation dans le réfrigérant, le solvant est récupéré dans le ballon de gauche. |
Basicité
Basicité de Lewis
L'existence de doublets non liants sur l'atome d'oxygène entraîne une réactivité de base de Lewis.
 |
Avec l'acide de Lewis BF3 on obtient un complexe stable : l'éthérate de trifluorure-bore.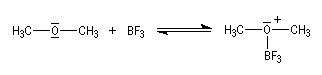 |
Ce caractère basique se manifeste vis à vis des organométalliques comme les magnésiens aussi bien à l'état solide qu'en solution. Dans ce cas plusieurs équilibres tels que l'équilibre de Schlenk peuvent intervenir simultanément.
Les éthers tels que Et2O manifestent des propriétés complexantes vis à vis de composés des éléments de transition.
 |
En ajoutant quelques gouttes d'une solution aqueuse de peroxyde d'hydrogène H2O2 (1 M) à une solution de dichromate de potassium K2Cr2O7 (1 M) acidifiée par H2SO4, on obtient de façon fugace une coloration bleue. En ajoutant de l'éthoxyéthane au mélange la coloration se stabilise. Après décantation, la phase éthérée est bleue. Elle contient un composé peroxo du chrome complexé par l'éther : [CrO(O2)2Et2O]. Remarque : les composés du Cr (VI) sont dangereux (caractère cancérigène). Porter des gants pour manipuler ces composés et travailler sous une hotte ventilée. |
Les éthers sont aussi des bases de Brönsted très faibles. Leurs acides conjugués constituent des sels d'oxonium qui sont des acides forts : pKa (R2OH+/R2O) # -3,5.
Le chlorure d'hydrogène est ionisé dans l'éthoxyéthane. Mais, à la différence de ce qui se passe dans l'eau, les solutions obtenues ne sont pas conductrices du courant électrique car l'éther, qui possède une constante diélectrique assez faible, (er = 4,3) si on la compare à celle de l'eau, (er = 78,5) ne permet pas la dissociation des paires d'ions.
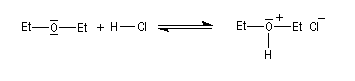
Coupure C-O
Action des acides
Les éthers, autres que les époxydes, sont stables en milieu basique sauf en présence de bases très fortes comme les organolithiens. En revanche, la liaison C-O peut être rompue en milieu acide, notamment par les acides halohydriques. Un réactif utile pour couper les éthers est HI qui les clive dès la température ambiante (réaction de Zeisel). Son rôle est double : H+ protone l'oxygène ce qui améliore considérablement son aptitude nucléofuge et I-, qui est un bon nucléophile, induit la coupure de la liaison C-O en réalisant une substitution nucléophile.
HBr coupe les éthers vers 100 °C et HCl à une température encore plus élevée car les anions sont de moins en moins nucléophiles.
Lorsque l'atome de carbone impliqué dans la fonction éther est plus substitué, on peut observer des substitutions de type SN1 qui s'accompagnent alors d'un certain pourcentage d'élimination.
 |
L'oxyde de ditertiobutyle est instable en milieu acide à
cause de la répulsion des groupes méthyle (examiner le modèle
"spacefill"). Comme la réaction passe par l'intermédiaire d'un carbocation, on obtient aussi une proportion non négligeable de méthylpropène par élimination.
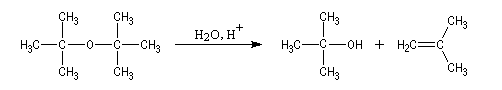 |
Du fait de leur faible réactivité, les éthers sont largement utilisés comme groupement protecteur des fonctions alcool et phénol. L'éther est formé à partir de l' alcoolate ou du phénolate au moyen de la réaction de Williamson. Comme il a été dit plus haut, la déprotection peut être réalisée en présence d'acide iodhydrique mais dans des conditions assez vigoureuses.
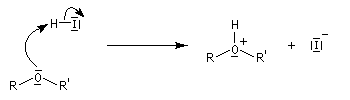
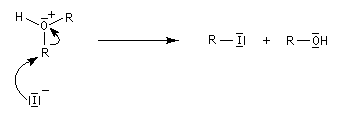
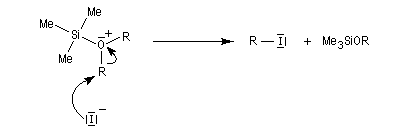
L'iodure de triméthylsilyle est un réactif très utile permettant la coupure des éthers dans des conditions douces.
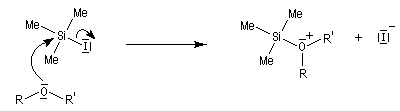
Les éthers de benzyle sont clivés sélectivement dans des conditions douces par H2 en présence de Pd/C.
L'utilisation des éthers silylés en tant que groupes protecteurs fait l'objet d'un paragraphe particulier.
Réactions au voisinage de la fonction éther
Autoxydation
On sait depuis longtemps que les éthers laissés au contact de l'air en présence de lumière peuvent conduire à des mélanges dangereusement explosifs.
La réaction entre l'éther et le dioxygène s'appelle autoxydation. Elle conduit à la formation d'hydroperoxydes (appelés quelquefois "peroxydes d'éther") selon un mécanisme radicalaire en chaîne initié par la lumière. Les hydroperoxydes sont thermodynamiquement instables du fait de la faible énergie de la liaison O-O et cinétiquement labiles. Ils peuvent exploser sous l'action d'une élévation de température, ou d'un choc.
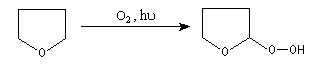
 |
Un test simple consiste à faire réagir la solution dans laquelle on soupçonne la présence d'hydroperoxyde avec une solution de KI (mélanger 0,5 mL d'éther, 1 mL de KI à 10 % et 0,5 mL d'acide éthanoïque. Agiter. En présence de peroxydes la solution devient brune car I-est oxydé en I2 qui forme avec I- l'ion I3- (image de gauche). L'expérience a été réalisée du THF provenant d'une bouteille entamée depuis quelques mois. Pour rendre le test plus sensible, on peut ajouter quelques gouttes d'un indicateur d'iode (empois d'amidon ordinaire ou Thiodène). Dans ce cas la solution devient bleue. |
Les éthers commerciaux, notamment le THF, sont stabilisés par des antioxygènes comme le 2,6-ditertiobutylparacrésol. Les phénols qui sont très sensibles à l'oxydation par O2 jouent le rôle de pièges à radicaux dans la réaction en chaîne d'oxydation. Le MTBE présente l'avantage par rapport aux autres éthers d'être très peu sensible à la peroxydation. Notons que les éthers ayant tendance à former des peroxydes, il ne faut pas les utiliser comme solvant dans les réactions impliquant des radicaux en présence d'oxygène.
Halogénation
La réaction avec Cl2 de type radicalaire, permet de substituer l'atome d'hydrogène porté par le C en a de l'atome d'oxygène par un atome de chlore.
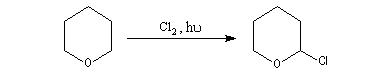
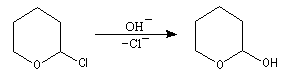
Elimination
Sous l'action de bases très fortes comme les organosodiques ou les organolithiens et dans une moindre mesure les organomagnésiens, l'atome d'hydrogène porté par le carbone en b de l'oxygène peut être arraché. Une réaction d'élimination conduit à un alcène et à un alcoolate.
Transposition de Claisen
Lorsqu'on chauffe à 200 °C le prop-2-ényloxybenzène (oxyde d'allyle et de phényle), on obtient le 2-(prop-2-ényl)-phénol (orthoallylphénol). La réaction constitue un exemple d'une réaction très générale qui affecte les éthers de vinyle et d'allyle : la transposition de Claisen mise en évidence par Ludwig Claisen (1851-1930).
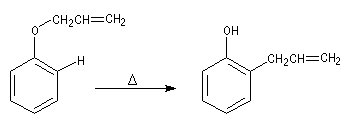

Introduction
Les époxydes ou oxacyclopropanes sont des éthers cycliques à trois chaînons. Le plus simple des époxydes est appelé oxacyclopropane ou oxyde d'éthylène. Les autres peuvent être considérés comme des dérivés substitués du précédent. Les époxydes sont des intermédiaires très souvent utilisés en synthèse organique où l'on met à profit leur grande réactivité. Ils combinent en effet l'intérêt de posséder une tension de cycle importante et d'avoir un atome d'oxygène nucléofuge. Ils peuvent être ouverts dans des conditions douces par de très nombreux réactifs en milieu basique et en milieu acide.
Synthèse des époxydes
Il existe plusieurs voies d'accès aux époxydes. En voici quelques-unes parmi les plus importantes.
- La préparation à partir d'un substrat éthylénique est traitée dans la partie consacrée à ces composés. Le réactif est un peroxoacide. La réaction est stéréospécifique de stéréochimie syn. Notons que l'époxydation de liaisons éthyléniques d'alcools allyliques peut être rendue énantiosélective en utilisant un complexe d'un métal de transition (Ti) et un tartrate comme auxiliaire chiral.
- A partir des composés carbonylés. Le réactif est un ylure de soufre. Cette réaction dite de Corey-Chaykovsky a acquis une grande importance en synthèse organique du fait de sa stéréosélectivité. Elle est examinée dans la partie consacrée aux composés carbonylés.
- Les halohydrines sont des précurseurs d'époxydes quand elles sont traitées en milieu basique par réaction de Williamson intramoléculaire.
- A partir des a-halogénoesters. La réaction entre un carbanion formé à partir d'un a-halogénoester et un composé carbonylé est appelée réaction de Darzens. Il s'agit d'une méthode de préparation d'époxyesters (esters glycidiques). Elle est étudiée dans le chapitre relatif aux esters.
Ouverture nucléophile par les ions hydroxyde
Le traitement par l'eau du plus simple des époxydes, l'oxacyclopropane, conduit à la formation d'éthanediol.
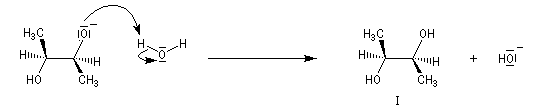
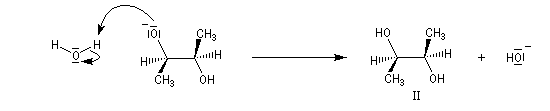
Dans les mêmes conditions, un mélange racémique d'époxydes (2R,3R)-diméthyloxacyclopropane et (2S,3S)-diméthyloxacyclopropane fournit le diol (2R,3S), c'est à dire un composé méso. Deux substrats diastéréo-isomères fournissent donc, dans les mêmes conditions des produits diastéréo-isomères. La réaction est donc diastéréospécifique.
Puisque les époxydes sont facilement obtenus par réaction entre un peroxyacide et un alcène, cette suite de réactions constitue une dihydroxylation de ceux-ci permettant la synthèse de diols. Si l'on se réfère à l'alcène de départ, l'ensemble des deux réactions est diastéréospécifique de stéréochimie anti. La dihydroxylation diastéréospécifique syn des alcènes est réalisable au moyen du permanganate de potassium ou du tétroxyde d'osmium.
Lorsque le cas se présente, l'ouverture nucléophile est régiosélective. Le nucléophile attaque l'époxyde sur l'atome de carbone le plus dégagé donc le moins substitué.
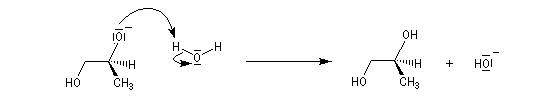
Ouverture nucléophile avec les organométalliques
La réaction entre un époxyde comme l'oxacyclopropane et un organomagnésien ou un organolithien constitue une synthèse d'alcool avec allongement de chaîne carbonée.
La réaction s'effectue en deux étapes distinctes :
- addition du réactif organométallique à l'époxyde conduit à un alcoolate magnésien ;
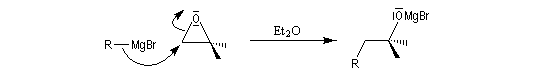
- cet alcoolate magnésien est ensuite hydrolysé en milieu acide (NH4Cl ou H2SO4 2 M) afin d'éviter la précipitation de Mg(OH)2.
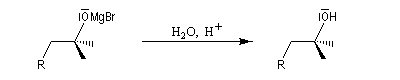
Autres ouvertures nucléophiles
Les réactions suivantes sont utiles en synthèse :
- alcoolates
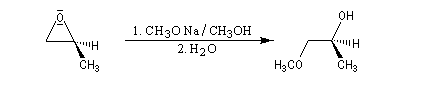
- hydrures (LiAlH4)
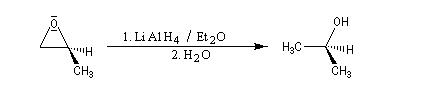
- ions alcynures
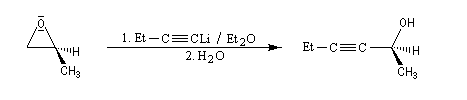
La déprotonation d'époxydes au moyen de bases fortes comme le LDA, permet la synthèse d'alcools allyliques.
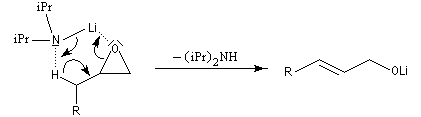
Il s'agit d'une réaction de transposition des 2,3-époxyalcools sous l'action d'une base [35].
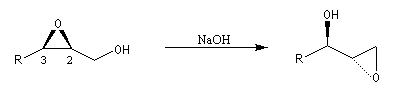
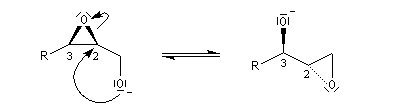
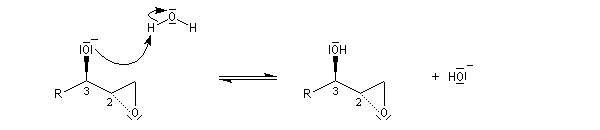
Ouverture énantiosélective
Certains complexes métalliques utilisant un ligand chiral dérivant du ligand salen ont été préparés assez récemment par E. Jacobsen (Harvard).
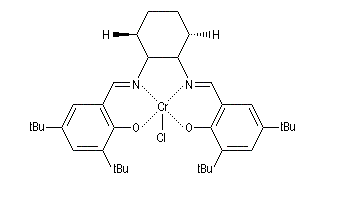
L'ouverture des époxydes méso peut être réalisée par déprotonation énantiosélective à l'aide d'une base chirale. Ce qui conduit à des alcools allyliques chiraux.
Ouverture des époxysilanes
 |
Un époxysilane peut être préparé par réaction entre un vinylsilane et un agent d'époxydation comme le m-CPBA. |
La réaction est régiosélective. L'attaque du nucléophile s'effectue sur l'atome de carbone qui stabilise le mieux une charge positive c'est à dire sur l'atome de carbone substitué par le plus grand nombre de groupes alkyles. On a donc une régiosélectivité inverse de ce qu'on observe en milieu basique. Avec un atome de carbone secondaire, la réaction reste bimoléculaire et stéréosélective anti.
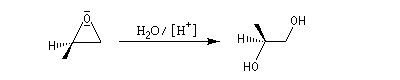
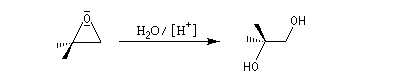
Les époxydes réagissent avec le trifluorure de bore BF3 pour donner des composés carbonylés.
L'atome d'oxygène, basique de l'époxyde réagit avec l'acide de Lewis BF3
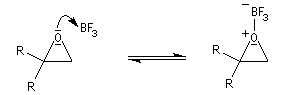

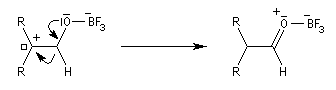
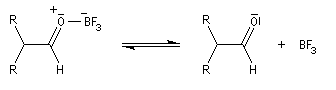
Préparation
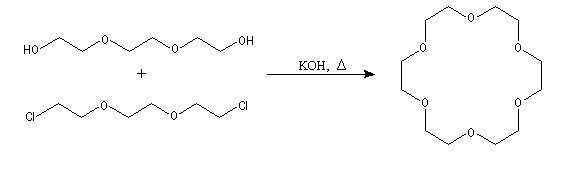
Le chauffage d'un mélange de 1,8-dichloro-3,6-dioxaoctane et de triéthylèneglycol en présence d'hydroxyde de potassium, conduit par une double réaction de Williamson à la synthèse d'un macrocycle possédant 18 chaînons. Ce composé est le représentant le plus connu d'une famille de composés préparés en 1967 par le chimiste C. J. Pedersen : les éthers-couronnes.
C. J. Pedersen (du Pont de Nemours), D. J. Cram (UCLA), J. M. Lehn (Université de Strasbourg) on obtenu le Prix Nobel de chimie en 1987 pour leurs travaux concernant les ligands macrocycliques : éthers-couronnes et macrobicycliques : cryptands.
|
L'image de gauche représente l'éther couronne [18]O6.
Dans la nomenclature internationale on le nomme : 1, 4, 7, 10, 13,
16-hexaoxacyclooctadécane. On peut le considérer comme un dérivé
cyclique de l'éthane-1,2-diol (éthylèneglycol). A la température
ordinaire ce composé se présente sous la forme d'un solide blanc (TF : 37,5 °C). Attention : les éthers couronnes sont des composés toxiques en raison de leur action sur le système nerveux. Ils doivent être manipulés avec précaution. Il faut porter des gants et travailler sous une hotte ventilée. |
![[18]O<SUB>6</SUB>](http://www.faidherbe.org/site/cours/dupuis/images5/18c6.gif)
Influence de la taille de la couronne
Le diamètre de la cavité de [18]O6 est compris entre 0,26 nm et 0,32 nm. Ce ligand hexadente est une base de Lewis qui forme facilement des complexes avec les ions suivants qui jouent le rôle d'acides de Lewis :
|
Ion
|
Rb+
|
NH4+
|
K+
|
|
d/nm
|
0,294
|
0,286
|
0,266
|
Transfert de phase
L'expérience suivante illustre la possibilité de transférer des ions en phase organique au moyen de [18]O6.
 |
 |
 |
|
I
|
II
|
III
|
L'ion K+ couronné est une nouvelle entité de grande dimension qui se comporte comme une molécule dans sa partie périphérique et comme un ion dans sa partie centrale. Ce complexe peut attirer MnO4- par interaction électrostatique tout en conservant une aptitude à se dissoudre en phase organique. K+ couronné permet le transfert de MnO4- de la phase aqueuse à la phase organique. La couleur du mélange évolue ensuite lentement vers le brun qui signale la présence de MnO2 . L'ion MnO4- qui est devenu très oxydant attaque la chaîne latérale du toluène et l'oxyde en acide benzoïque.
Il existe d'autres agents de transfert de phase :
- les ions ammonium quaternaires ;
- les cryptands.
Dans un solvant protique (eau, alcool etc.) les anions sont fortement solvatés car ils contractent avec le solvant des liaisons hydrogène. L'intensité de cette solvatation est d'autant plus importante que l'ion est plus petit et plus chargé.
La nucléophilie est l'aptitude d'une base de Lewis à donner son doublet. Il s'agit d'un concept cinétique. On peut admettre raisonnablement que la couche de solvant entourant un anion solvaté camoufle son cortège électronique externe. C'est, en partie la raison pour laquelle dans ce type de solvants la réactivité nucléophile des anions varie dans l'ordre : I-> Br- > Cl- > F-. Ainsi, il n'est généralement pas possible d'obtenir des composé fluorés par substitution nucléophile bimoléculaire à partir d'un bromure d'alkyle comme substrat même en présence d'un gros excès de KF.
Remarque : en vertu du principe HSAB de Pearson on peut aussi dire que la base dure F- interagit fortement avec l'acide dur S-H constitué par le solvant protique donneur de liaison H.
En revanche la réaction entre le bromo-1-octane et l'ion fluorure s'effectue avec un rendement dépassant 90 % en présence d'éther-couronne [18]O6.
|
Composé
|
I
|
II
|
|
%
|
92
|
8
|
Ethers d'énols
L'exemple du dihydropyrane (DHP)
Les éthers d'énols sont des composés bifonctionnels qui combinent la fonction éther avec une insaturation en a de l'atome d'oxygène. Le dihydropyrane est un réactif utile facile à préparer et de coût peu élevé.
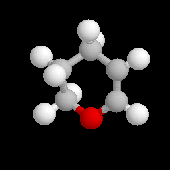 |
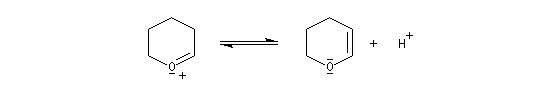
Le DHP peut être obtenu à partir du 2-hydroxypyrane par une réaction d'élimination en milieu acide.
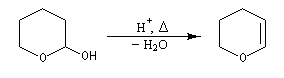 |
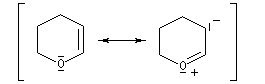
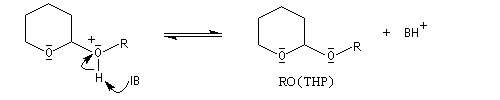
Le DHP est largement utilisé comme groupe protecteur de la fonction alcool. On utilise une quantité catalytique d'un acide tel que l'APTS. L'acétal formé noté RO(THP) est stable en milieu basique et inactif vis à vis de nombreux réactifs tels que les organométalliques ou LiAlH4. L'exemple suivant illustre l'utilisation de la protection par le DHP dans une synthèse magnésienne.
- protection de l'alcool par réaction avec le DHP en
catalyse acide pour conduire à un éther de tétrahydropyranyle qui est en
fait un cétal du fait de la présence d'un atome d'oxygène sur le cycle ;
le mécanisme de la protection est le suivant.
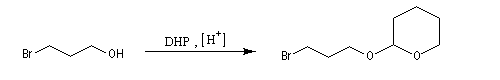
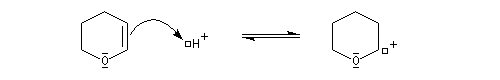
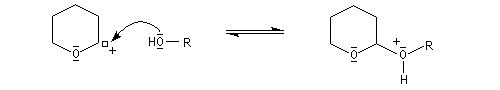

- synthèse du magnésien ;
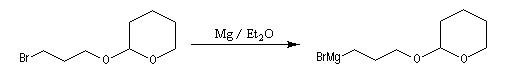
- réaction entre l'organomagnésien et le composé carbonylé suivie de l'hydrolyse de l'alcoolate magnésien en milieu faiblement acide (NH4Cl) ;
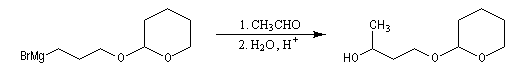
- déprotection de la fonction alcool par hydrolyse en milieu acide ;
le mécanisme de la déprotection est donné ci-dessous.
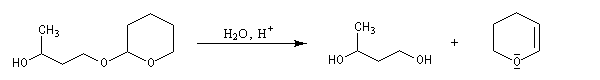
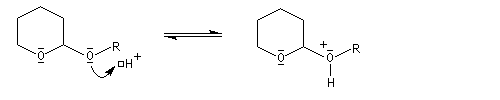
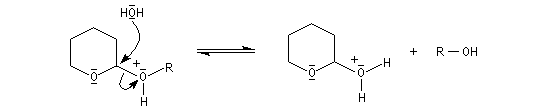
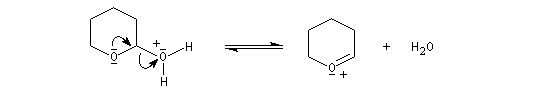
 |
La montmorillonite K 10 est une argile qui se présente sous la forme d'une poudre de couleur grise de granulométrie très fine et de grande surface spécifique (220-270 m2.g-1). C'est un catalyseur acide qui présente plusieurs propriétés remarquables : La structure en feuillets des argiles permet une accélération importante de la réaction car les molécules se déplacent dans un plan et non dans l'espace ce qui accroît leur probabilité de rencontre. D'un point de vue technique, le produit étant adsorbé sur l'argile, il est facile de le séparer du mélange réactionnel par simple filtration. |
Furane et dérivés substitués
Le furane et ses dérivés substitués peuvent être considérés comme des éthers d'énols. L'hydrolyse en milieu acide conduit à une ouverture du cycle.
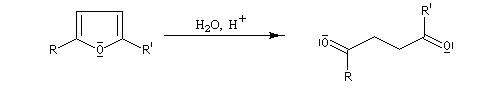
Synthèse
Dans un éther de silyle, le silicium remplace le carbone dans sa liaison avec l'oxygène. Le tableau suivant regroupe quelques valeurs d'énergies de liaison, qui illustrent la grande énergie de la liaison Si-O si on la compare à celle de la liaison C-O. L'origine de cette valeur élevée est à rechercher dans la différence d'électronégativité importante entre les atomes de silicium et d'oxygène et à la polarisabilité du silicium plus importante que celle du carbone. Cette propriété est à mettre en relation avec le fait que le silicium possède un rayon atomique plus élevé que l'atome de carbone.
|
liaison
|
C-O
|
Si-C
|
Si-O
|
|
Do / kJ.mol-1
|
343
|
372
|
531
|
Les éthers de silyle sont très utilisés comme groupe protecteurs des fonctions possédant un atome d'hydrogène acide alcools, phénols, acétyléniques. L'un de leur intérêt réside dans leur plus grande volatilité que les éthers ordinaires ce qui permet une purification facile en chromatographie. Ils sont également utilisés en spectrométrie de masse dans la détermination des masses molaires. On les prépare généralement par réaction entre l'alcool et un chlorure de trialkylsilyle.
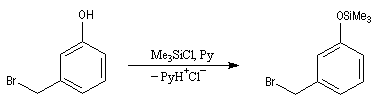
|
triméthylsilyle
|
t-butylsilyle
|
t-butyldiméthylsilyle
|
t-butyldiphénylsilyle
|
|
TMS
|
TBS
|
TBDMS
|
TBDPS
|
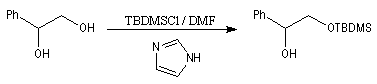
Le clivage des éthers de silyle peut s'effectuer de plusieurs façons :
- substitution nucléophile sur le silicium par CH3Li en présence de LiBr ;
- substitution nucléophile par les ions F- sur le silicium, réaction qui met cette fois à profit la grande énergie de liaison Si-F (582 kJ.mol-1). Un réactif intéressant pour effectuer cette transformation est le fluorure de tétrabutylammonium (TBAF) introduit par Corey. Le mécanisme fait intervenir un intermédiaire dans lequel le silicium est pentacoordiné (mécanisme SN2-Si).
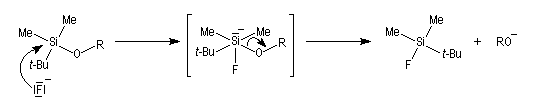
Ethers d'énols silylés
Préparation
Les éthers d'énols silylés sont des intermédiaires de réaction qui ont acquis une grande importance dans les réactions organiques modernes. L'énolate, obtenu par réaction entre le composé carbonylé et une base forte comme le LDA est piégé sous forme d'éther d'énol par réaction avec le chlorure de triméthylsilyle en présence de Et3N. Un réactif permettant d'effectuer la réaction dans des conditions très douces est l'iodure de triméthylsilyle en présence de (Me3Si)2NH.
 |
Un exemple de préparation d'éther d'énol silylé est donné ci-dessous.
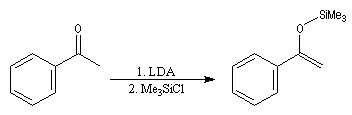 |
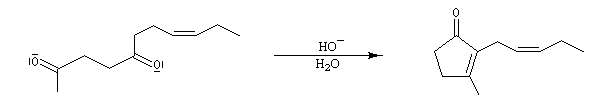
La cible recherchée est la 2-bromo-6-méthylcyclohexanone P. La bromation directe de la 2-méthylcyclohexanone fournit un mélange d'isomères avec une mauvaise régiosélectivité. L'idée est de préformer l'énolate qui doit réagir. Une base encombrée et très forte comme le LDA arrache l'atome d'hydrogène le plus accessible et fournit l'énolate cinétique E.
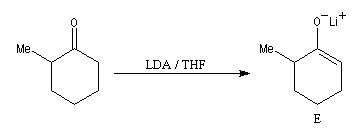
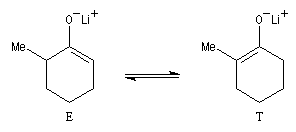
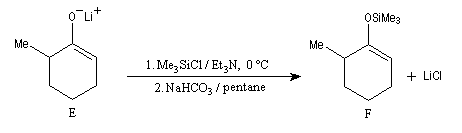
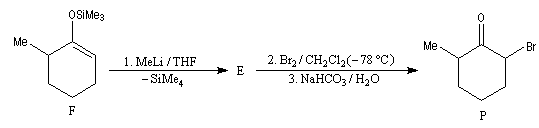
Les éthers d'énols silylés peuvent subir un clivage oxydant sous l'action de la dichlorodicyanoquinone (DDQ). On prépare ainsi des a-énones qui sont des intermédiaires de synthèses précieux [30].
L'exemple suivant concerne la synthèse de la carvone (Fleming et Paterson, 1979) [31].
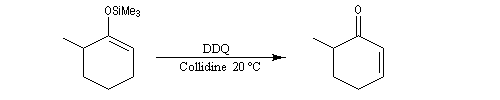
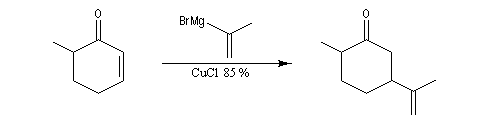
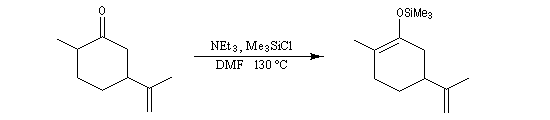
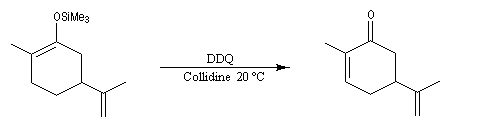
On commence par préparer l'énolate cinétique de la méthylcyclohexanone. Ce dernier est ensuite piégé sous forme d'éther d'énol silylé. L'énolate réagit au niveau de l'oxygène avec le chlorure de triméthylsilyle (exemple d'interaction acide dur-base dure).
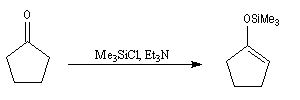
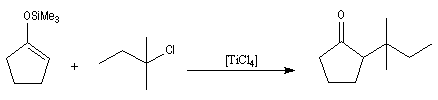
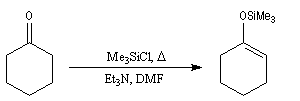
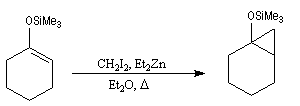
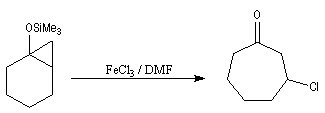
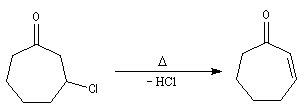
La réaction de Mukaiyama constitue un type particulier de condensation aldolique entre un éther d'énol silylé et un aldéhyde en présence d'un acide de Lewis comme TiCl4.
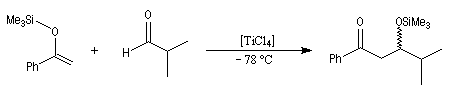
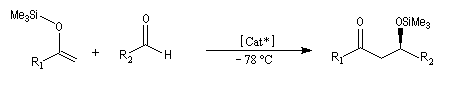
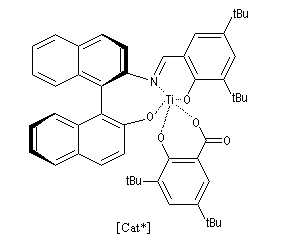
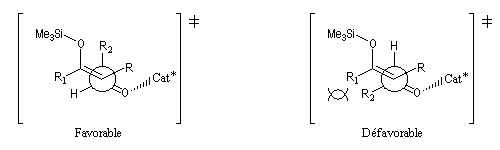
L'état de transition favorisé répond à deux critères :
- l'addition nucléophile de l'énol s'effectue préférentiellement sur la face prochirale la plus dégagée de l'aldéhyde donc du côté opposé au catalyseur chiral ;
- l'éther d'énol est disposé de manière à réduire les interactions stériques.
Certains composés d'intérêt biologique comportent une fonction éther associée ou non à d'autres fonctions.
Safrole
 |
Le safrole est l'un des constituants principaux de l'essence de sassafras |
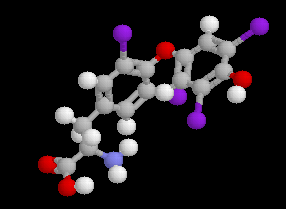 |
La thyroxine, hormone extraite de la glande thyroïde, fut découverte en 1920 par Kendall. Il s'agit d'une molécule chirale. La synthèse de la thyroxine racémique par iodation de la thyrosine est réalisée par Harington en 1927. En 1930, Harington et Salter ont identifié la thyroxine naturelle comme étant la forme L. La synthèse de la L-thyroxine a été réalisée par Chalmer en 1949. |
Le disparlure est une phéromone du bombyx disparate. La synthèse asymétrique de l'énantiomère (+) a constitué l'une des premières grandes applications de l'époxydation énantiosélective de Sharpless.
Bibliographie
Ouvrages généraux
J. March - Advanced Organic Chemistry (J. Wiley).
N. T. Anh - Introduction à la chimie moléculaire - Cours de l'Ecole Polytechnique (Ellipses 1994).
P. Laszlo - Logique de la synthèse organique - Cours de l'Ecole Polytechnique (Ellipses 1993)
R. Brückner - Mécanismes réactionnels en chimie organique (De Boeck 1999).
Ouvrages axés sur la partie expérimentale
M. Chavanne, A. Jullien, G. J. Beaudoin, E. Flamand - Chimie organique expérimentale (2ed, 1991 - Modulo).
L. F. Fieser et K. L. Williamson (fifth edition 1983 D. C. Heath ans Co)
Liens
[18] The Nobel Prize in Chemistry 1987
[18] purification du THF
[18] 18-Crown-6 by George W. Gokel, Donald J. Cram, Charles L. Liotta, Henry P. Harris, and Fred L. Cook.
[18] o-Eugenol by C. F. H. Allen and J. W. Gates, Jr.
[18] Ring expansion of Cycloalkanone by Yoshihiko Ito, Shotaro Fujii, Masashi Nakatuska, Fumio Kawamoto, and Takeo Saegus
[18] a-tert-alkylation of ketones by M. T. Reetz, I. Chatziiosifidis, F. Hübner, and H. Heimbach
[18] Désymétrisation d'époxydes Thèse M. P. Nury
[18] Introduction à la chimie supramoléculaire Teodor Silviu Balaban, Université d' Aix-Marseille III, „Paul Cezanne“; Karlsruhe Institute of Technology
[18] The Discovery of Crown Ethers, Nobel Lectures 1987 by Ch. Pedersen
[18] Epoxide Migration (Payne rearrangment) and related reactions by M. Manson.
[18] Mukayama Aldol reaction by Lu Kui, 2005.
[18] Protection et déprotection Ecole Polytechnique.
Articles
[30] I. Paterson, C.J. Cowden, V.S. Rahn, M.D Woodrow, Synlett 8, 915-917, Thieme, 1998
[31] I. Paterson, I. Fleming, Synthesis 1979, 736.
[32] T. Mukaiyama et al, J. Am. Chem. Soc. 96, 7503 (1974).
[33] E. Carreira, J. Am. Chem. Soc. 1994, 116, 8837.
[34] A. Gorgues - Activation anionique à l'aide d'ammonium quaternaires, d'éthers couronnes et de dérivés analogues BUP 595, Juin 1977.
[35] Payne, G. B. J. Org. Chem. 1962, 27, 3819.
[36] Chalmers et al., J. Chem. Soc. 1949, 3424.
Aucun commentaire:
Enregistrer un commentaire