Cours de chimie organique -
Benzène et composés aromatiques
Le benzène
Origine et structure
Le benzène est un hydrocarbure de formule brute C6H6, liquide à la température ordinaire, facile à cristalliser à 5,5 °C, d'indice de réfraction élevé, voisin de celui du verre.
 |
Dans la bouteille photographiée ci-contre, se trouve un tube scellé qui contient le premier échantillon de benzène isolé par M. Faraday dans le gaz d'éclairage. Cet échantillon est conservé dans le musée de la Royal Institution de Londres [45]. En 1834 E. Mitscherlich l'a préparé en chauffant de l'acide benzoïque en présence de chaux et lui a donné son nom. Le portrait est celui de Kathleen Lonsdale qui détermina en 1929 la structure précise du benzène par diffraction des rayons X. (photographie G. D. 2011) |
| La structure du benzène a été déterminée en 1929 par
diffraction des rayons X (voir plus haut.) Les 6 atomes de carbone
occupent les sommets d'un hexagone régulier. Les longueurs de liaison
entre atomes de carbone sont toutes égales à 0,140 nm, intermédiaire
entre une liaison simple (0,154 nm) et une liaison double (0,134 nm).
Les 6 atomes d'hydrogène sont dans le même plan que les 6 atomes de
carbone. On raconte que Kékulé trouva la formule cyclique du benzène en
1865 en rêvant à un serpent qui se mordait la queue. Il peut être obtenu
par trimérisation de l'éthyne.
Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
Formules du benzène
Plusieurs formules ont été proposées pour rendre compte de la structure du benzène. Le prismane de Ladenburg correspond à la formule pondérale mais laisse prévoir l'existence de quatre isomères dibromés alors que l'expérience montre qu'il n'en existe que trois. Les composés III et III' sont énantiomères.

En 1865 Kékulé proposa une formule cyclique formée par l'alternance de liaisons simples et doubles. Un tel système est appelé système conjugué. Dans cette optique, le benzène est le cyclohexa-1,3,5-triène. Cette formule suggère l'existence de 2 dérivés dibromés sur les atomes de carbone 1 et 2 mais l'expérience ne permet d'en mettre qu'un seul en évidence. De plus elle suppose l'existence de liaisons simples et doubles alternées. Or on sait par ailleurs que ces liaisons possèdent des longueurs différentes. Cela ne permet pas d'interpréter l'existence d'un hexagone régulier.
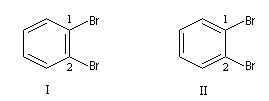
La méthode de la résonance appliquée au benzène Dans la méthode de la mésomérie, la molécule de benzène peut être représentée par les formes suivantes :
On interprète ainsi la longueur des liaisons dans la molécule (0,140 nm), intermédiaire entre celle d'une liaison simple (0,154 nm) et une liaison double (0,134 nm).
Dans la représentation de Robinson et Shortland, la délocalisation électronique des électrons est schématisée par un cercle à l'intérieur du cycle. Il est souhaitable de réserver cette représentation aux composés monocycliques.
Energie de résonance du benzène L'hydrogénation du benzène s'effectue en bloc et conduit au cyclohexane. Elle nécessite des conditions expérimentales assez vigoureuses et un catalyseur. On interprète la grande efficacité du nickel par le fait que le réseau cubique à faces centrées de ce métal est adapté à la géométrie hexagonale du benzène.
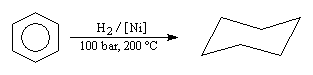
DrS0 << 0
On passe en effet de 4 mol de gaz dans l'état initial à une seule dans l'état final.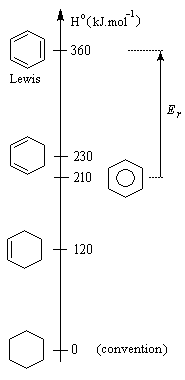 |
L’hydrogénation du benzène dégage une énergie de -210 kJ.mol-1. Celle du cyclohexène dégage -120 kJ.mol-1. On en déduit que l’hydrogénation du 1,3,5-cyclohexatriène (formule de Lewis) à trois doubles liaisons isolées fournirait : 3´ (-120) = -360 kJ.mol-1. L'énergie de cette structure hypothétique est donc supérieure de 360 kJ.mol-1 de celle du cyclohexane. L’énergie de résonance est la différence entre l’énergie de cyclohexa-1,3,5-triène (hypothétique) et celle de la molécule de benzène (réelle). C'est donc une grandeur positive.
ER = 360 - 210
ER = 150 kJ.mol-1
Cette quantité mesure donc l'accroissement de stabilité qu'acquiert la molécule de benzène du fait de son caractère aromatique. |
Hydrocarbures dérivés du benzène
Cumène
|
On peut préparer le cumène par réaction de Friedel et Crafts entre le benzène et le propène.
Dans l'industrie, la réaction s'effectue sous une pression de 35 bar à
une température de 190 °C en présence d'un catalyseur acide. L'oxydation du cumène par le dioxygène de l'air permet la préparation du phénol et de la propanone. Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
|
Le styrène existe à l'état naturel dans certains végétaux comme la
canelle. Dans l'industrie, on le prépare essentiellement par
déshydrogénation de l'éthylbenzène. Le
styrène est utilisé comme monomère dans la synthèse du polystyrène. Ce
dernier intervient dans de nombreuses matières plastiques
(film, polystyrène expansé, polystyrène choc). Il est aussi utilisé en
copolymérisation. Par exemple, la copolymérisation avec le buta-1,3-diène, permet de synthétiser des caoutchoucs synthétiques (SBR). Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
[p]-annulènes
On a bien sûr cherché à synthétiser les homologues inférieurs et supérieurs du benzène. Ces hydrocarbures sont des molécules conjuguées, cycliques, que F. Sondheimer a proposé d'appeler [p]-annulènes. La valeur de [p] donne le nombre de chaînons du cycle.
[p]
|
4
|
6
|
8
|
Nom
|
cyclobutadiène
|
benzène
|
cyclooctatétraène
|
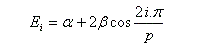
Les coefficients des orbitales moléculaires d'un [p]-annulène sont donnés par la relation ci-dessous.
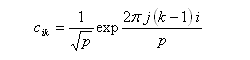
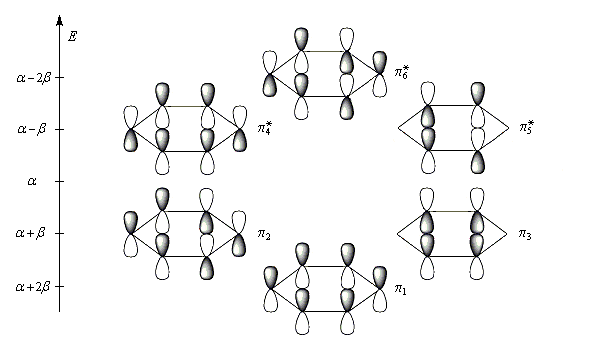
DE = 2b
Puisque DE < 0, la molécule de benzène est plus stable que celle de 1,3,5-cyclohexatriène (hypothétique). L'énergie de résonance électronique ER est par définition, l'opposé de cette grandeur :
Les niveaux d'énergie du cyclobutadiène (I), du benzène (II) et du cyclooctatétraène (III), prévus par la méthode de Hückel simple (HMO), sont donnés ci-dessous.
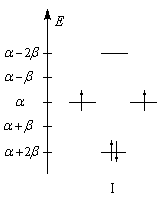 |
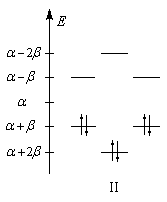 |
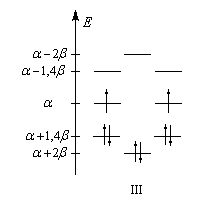 |
DE = 0
|
DE = 2b
|
DE = -1,65b
|
- b1 associée aux liaisons longues (simples) ;
- b2 associée aux liaisons plus courtes (doubles).
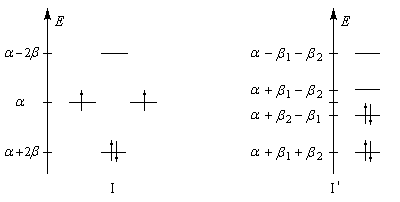
Le spectre infrarouge du cyclobutadiène a été réalisé à très basse
température (4 K) dans des matrices de gaz inerte (Ar ou Xe). On peut
représenter schématiquement la structure
par les formes mésomères suivantes : Représentation : spacefill ball & stick sticks wireframe |
| Le cyclooctatétraène ou [8]-annulène a été préparé
pour la première fois par R. Willstätter (Prix Nobel 1930) en 1911. La
méthode était basée sur la dégradation d'un composé naturel extrait de
la grenade : la pseudopelletiérine (nommée ainsi en hommage
au chimiste Pelletier qui l'a découverte). W. Reppe a montré en 1946 que le cyclooctatétraène pouvait également être obtenu par tétramérisation de l'éthyne (acétylène). Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
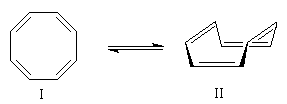
Comme on l'a dit, le benzène est un représentant d'une famille de [p]-annulènes particulièrement stables. En 1931 H. Hückel, a généralisé les résultats obtenus pour le benzène à d'autres hydrocarbures et a énoncé une règle permettant d'estimer la stabilité d'un [p]-annulène par comparaison avec un polyène conjugué acyclique. Les [p]-annulènes qui vérifient ce critère possèdent une stabilité particulière. On dit qu'ils sont aromatiques.
Un hydrocarbure est aromatique s'il est : monocyclique, plan et qu'il possède 4n + 2 électrons délocalisables.
Le benzène est un hydrocarbure aromatique qui correspond à la valeur n = 1.
Le cyclobutadiène est en revanche, le représentant d'une famille de [p]-annulènes particulièrement instables. R. Breslow a proposé d'appeler antiaromatiques ces composés pour rappeler qu'ils sont moins stables que les polyènes acycliques correspondants.
Un hydrocarbure est antiaromatique s'il est : monocyclique, plan et qu'il possède 4n électrons délocalisables.
Le cyclobutadiène (n = 1) et le cyclooctatétraène (n = 2) sont des hydrocarbures antiaromatiques.
Lorsqu'on utilise la règle de Hückel, il ne faut pas oublier l'une des trois conditions. Ainsi, un composé comme le barrelène possède, comme le benzène, 4n + 2 électrons p. Mais il n'est pas aromatique car ce n'est pas un annulène.
Vérifications de la règle de Hückel
Après le benzène, le premier annulène qui satisfait la formule de Hückel est le [10]-annulène. Il existe plusieurs diastéréoisomères qui peuvent adopter une infinité de conformations :
- la conformation plane du composé Z, Z, Z, Z, Z est un polygone régulier dont les angles de 144° révèlent une contrainte angulaire très importante ;
- le composé E, Z, Z, Z, Z est stable en conformation croisée non plane ;
- le composé Z, E, Z, Z, E est représenté ci-dessous.
| Dans le composé doublement trans représenté
ci-contre, il n'y a pas de contrainte angulaire mais la répulsion entre
les atomes d'hydrogène situés sur les atomes de carbone les plus proches
entraîne un gauchissement du cycle. Le composé n'est pas aromatique car
il n'est pas plan.
Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
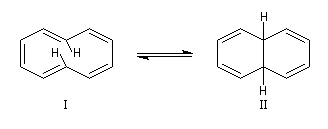
| L'hydrocarbure suivant qui satisfait la formule de Hückel
est le [14]-annulène. Ce composé est moins tendu que le précédent mais
il existe aussi sous deux formes en équilibre qui ne sont pas
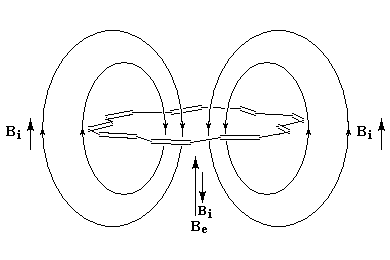
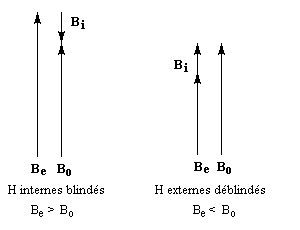
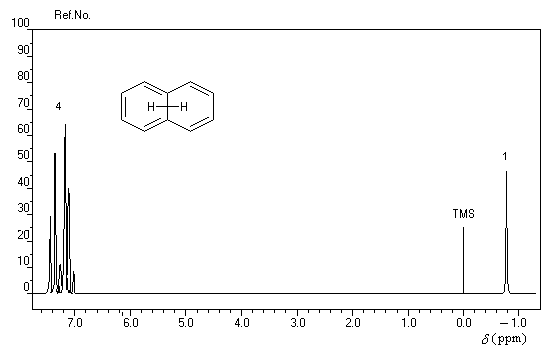
rigoureusement planes par suite des contraintes angulaires. La spectroscopie RMN montre l'existence d'un courant de cycle diamagnétique. Les protons externes sortent à d = 7,6 ppm tandis que les protons internes sortent à d = 0,0 ppm. Le [14]-annulène a donc un caractère aromatique.
Zoom :
in
out
Représentation : spacefill ball & stick sticks wireframe Atomes d'hydrogène internes : on off Atomes d'hydrogène externes : on off |
| Le [18]-annulène est un composé
de couleur rouge brun qui a été préparé par F. Sondheimer (University
College Londres) en 1962 en mettant à profit une réaction de couplage oxydant
des alcynes (couplage de Eglinton). L'étude radiocristallographique aux
rayons X montre que la molécule de [18]-annulène est plane. L'énergie
de résonance est voisine de celle du benzène : ER = 154 kJ.mol-1. Zoom : in out Représentation : spacefill ball & stick sticks wireframe Atomes d'hydrogène internes : on off Atomes d'hydrogène externes : on off |
Hydrocarbures polycycliques
Il existe de nombreux hydrocarbures possèdant des noyaux benzéniques accolés. La parenté de ces composés avec le benzène entraîne pour certains d'entre-eux un caractère aromatique indéniable (réactions de substitution électrophiles etc). Cependant il faut faire attention que la règle de Hückel, qui n'est valable que pour les hydrocarbures monocycliques, ne s'applique pas à ces hydrocarbures (en ce qui concerne le mode de représentation, il est préférable de réserver aux composés monocycliques, la notation avec un cercle inscrit dans le cycle).
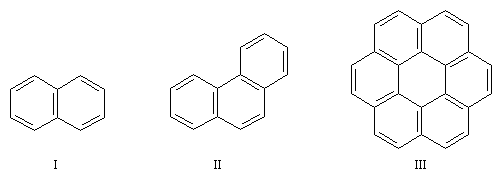
Composé
|
naphtalène (I)
|
phénantrène (II)
|
coronène (III)
|
électrons p
|
10
|
14
|
24
|
| Le coronène possède 24 électrons p. La molécule est plane et possède un caractère aromatique incontestable. Le coronène ne satisfait pas la formule de Hückel. Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
En ce qui concerne les composés polycycliques, les cycles ne possèdent pas tous le même caractère aromatique. Ainsi la réactivité du cycle central de l'anthracène le rapproche d'un diène conjugué comme en témoignent : son oxydation facile en anthraquinone et la réaction de Diels-Alder avec le tétracyanoéthène (TCNE).
Buckminsterfullèrene
| La molécule de buckminsterfullerène possède 62 électrons p.
Elle a été élue molécule de l'année 1991 par le magazine Science. Bien
qu'elle comporte des cycles de type benzénique, la molécule n'est pas
aromatique car elle n'est pas plane. Sa réactivité est bien plus grande
que celle du benzène. Ses découvreurs, Robert F. Curl, Sir Harold W.
Kroto, Richard E. Smalley ont obtenu le prix Nobel de chimie en 1996 [36]. (modèle par D. Woodcock) Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
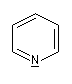
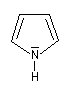
Présentation Un hétérocycle possède un ou plusieurs hétéroatomes à l'intérieur d'un cycle d'atomes de carbone. Le tableau suivant regroupe quelques hétérocycles simples.
 |
 |
 |
 |
pyridine
|
pyrrole
|
furane
|
thiophène
|
Dans la pyridine le doublet non liant de l'azote ne participe pas au système aromatique. En revanche dans le pyrrole ce doublet y participe.
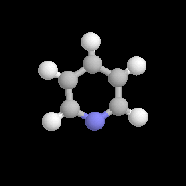 |
Sous 1 bar, la pyridine est un liquide (TE = 115
°C). Le doublet sur l'azote ne participe pas au système aromatique.
C'est la raison pour laquelle la pyridine a des propriétés basiques : pKa (PyH+/PyH)
= 8,6. La pyridine est un bon solvant de nombreux composés organiques
elle est également miscible à l'eau (grâce aux liaisons H).
C'est un composé toxique dont l'odeur est particulièrement écœurante. On l'utilise fréquemment comme piège à protons et comme catalyseur nucléophile dans l'acylation des alcools et des amines. |
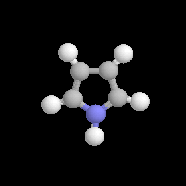 |
Sous 1 bar, le pyrrole est un liquide (TE = 129 °C). Contrairement à la pyridine, le doublet de l'azote participe au système aromatique. De ce fait, le pyrrole est une base très faible : pKa (PyrH+/PyrH) = 0,4. Le cation n'a aucun caractère aromatique. Le pyrrole est aussi un acide très faible qu'on peut déprotoner grâce à un métal alcalin comme le potassium. pKa (PyrH/Pyr-) = 16,5. L'anion pyrrolique est, comme le pyrrole, un système aromatique. Il se comporte comme un nucléophile réactif car le doublet peut être cédé tout en conservant son caractère aromatique. |
Les porphyrines sont des composés aromatiques polycycliques dans lesquels on trouve quatre unités pyrrole. Ce système plan qui comporte 18 électrons délocalisés, peut être rapproché du [18]-annulène. L'hème est un complexe porphyrine-fer (II) qu'on trouve dans plusieurs protéines notamment la myoglobine et l'hémoglobine.
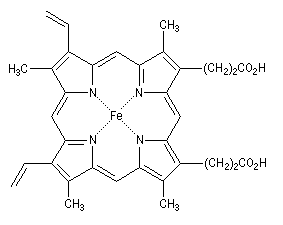 |
L'hème représentée à gauche est présente dans plusieurs protéines :
|
Cation tropylium
La structure du carbocation cyclohepta-1,3,5-triénium (cation tropylium) a été établie par Doering en 1954 [58]. Cette détermination a eu un assez grand retentissement à l'époque car elle constituait l'une des premières vérification de la règle de Hückel. Une préparation est décrite à la référence [35].
Un autre exemple remarquable de carbocation aromatique est le cation cyclopropénium qui, bien que présentant des contraintes stériques importantes, a pu être préparé par R. Breslow en 1967.
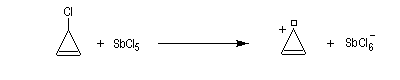
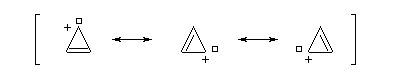
Anion cyclopentadiényle L'expérience montre qu'il existe une grande différence d'acidité entre le méthane (pKa = 60) et le cyclopentadiène (pKa = 20). L'anion cyclopentadiényle est facilement préparé par réaction entre le cyclopentadiène et l'hydroxyde de sodium dans le DMSO. Ce solvant dipolaire aprotique exalte la basicité de l'ion OH- en solvatant très efficacement le contre-ion K+.
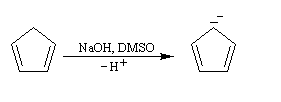
L'ion cyclopentadiényle est un carbanion stabilisé grâce à la présence d'un système conjugué qui peut être décrit en utilisant la méthode de la mésomérie.
Le ferrocène L'année 2001 a marqué le cinquantième anniversaire de la publication par P. L. Pauson et T. J. Kealy de la formation d'un complexe du fer dont la structure et les propriétés sont remarquables : le ferrocène [55], [56]. On le prépare assez facilement par réaction entre l'ion cyclopentadiényle et les ions Fe2+ dans le diméthylsulfoxyde (DMSO) [3].
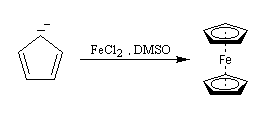
- après déprotonation du cyclopentadiène par réaction d'oxydo-réduction avec le sodium dans le xylène, l'ion cyclopentadiényle est mis à réagir avec le chlorure de fer (II) dans le THF ;
- une amine suffisamment basique comme la diéthylamine peut être utilisée pour former l'ion cyclopentadiényle à partir du cyclopentadiène.
 |
A la température ordinaire, le ferrocène se présente sous la forme d'un solide (TF = 175 °C) de couleur orange possédant une grande stabilité chimique. Préparé pour la première fois en 1951 par P.L. Pauson, ce composé a excité l'imagination des chimistes en raison du mode original de liaison entre les cycles et le fer. Il est le chef de fil d'une famille de composés appelés métallocènes. L'étude de ces composés a été menée activement dans les années 50 par deux équipes indépendantes sous la direction de G. Wilkinson (Harvard) et E. O. Fischer (Munich). Ce travail leur a valu le prix Nobel en 1973 [32]. |
- un moment dipolaire nul ;
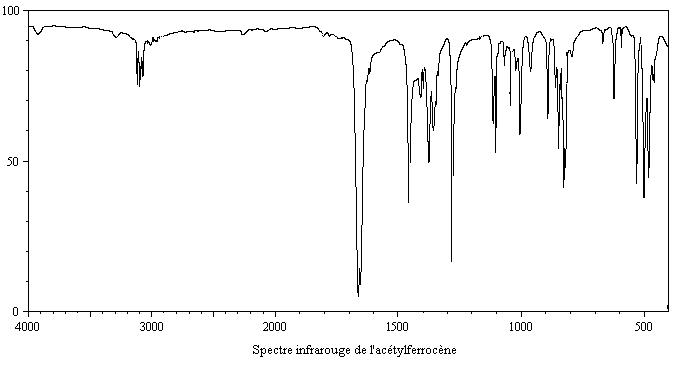
- en spectroscopie infrarouge, un petit nombre de bandes d'absorption dans la région 650 cm-1 - 4000 cm-1, ce qui témoigne d'une structure très symétrique, avec en particulier une bande d'absorption à 1410 cm-1 caractéristique d'un cycle cyclopentadiényle non substitué [31] ;
- un caractère diamagnétique.
| Le ferrocène est un complexe de type p
dans lequel les cycles se situent au dessus et en dessous de l'ion Fe
(II). L'analyse par diffraction des rayons X et par diffraction des
neutrons montre que la conformation
la plus stable est celle dans laquelle les deux cycles sont
pratiquement éclipsés avec un angle entre les deux cycles est voisin de
9° (symétrie D5h). La conformation dans laquelle les cycles sont décalés (symétrie D5d) possède une énergie supérieure d'environ 4 kJ.mol-1. Le fichier pdb du ferrocène m'a été fourni par le Dr R. J. Lancashire que je remercie particulièrement.
Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
Spectroscopie
Spectroscopie UV
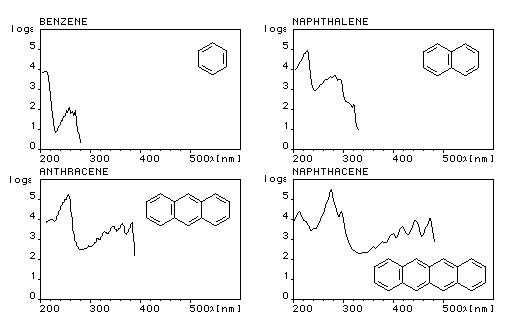
Le spectre UV du benzène présente trois bandes d'absorption :
- Bande E : lm = 185 nm, intense e = 50000
- Bande E : lm = 204 nm, moyenne e = 7000 (épaulement de la précédente)
- Bande B : lm = 260 nm, faible e = 200 (possède une structure fine)
Les spectres UV des polyacènes ressemblent à celui du benzène mais les longueurs d'onde des maxima d'absorption sont déplacés vers les grandes longueurs d'onde (effet bathochrome) et l'intensité des bandes est accrue.
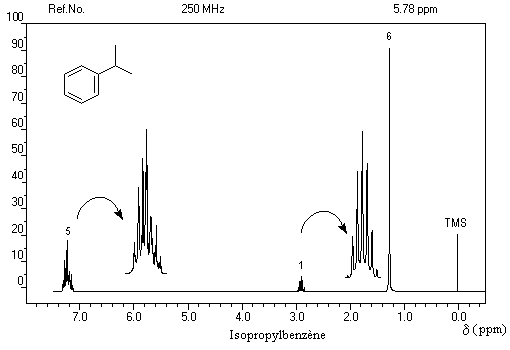
- d = 1,2 ppm doublet : 6 H des groupes méthyle ;
- d = 3 ppm septuplet : 1 H du groupe isopropyle ;
- d = 7,3 ppm multiplet complexe : protons du cycle aromatique.
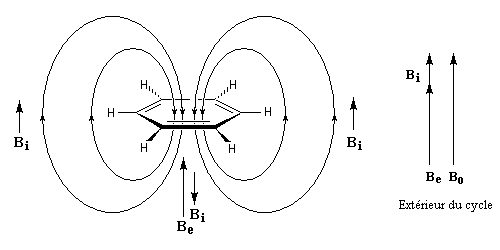
- l'aromaticité entraîne un déplacement chimique des protons externes vers les champs faibles dû à l'existence d'un courant de cycle diamagnétique. Un composé aromatique est qualifié de diatropique ;
- l'antiaromaticité, en revanche, entraîne un déplacement chimique des protons externes vers les champs forts dû à l'existence d'un courant de cycle paramagnétique.
Le dessin suivant se rapporte au [18]-annulène mais peut être généralisé à d'autre [p]-annulènes.
 |
 |
Composé
|
ortho (3J)
|
méta (4J)
|
para (5J)
|
J (Hz)
|
7-8
|
2-3
|
0-1
|
Composé
|
 |
 |
 |
Signal RMN
|
2 massifs
|
3 massifs
|
1 massif
|
Remarque : dans l'exemple ci-dessous, noter le signal du proton aldéhydique très déblindé.
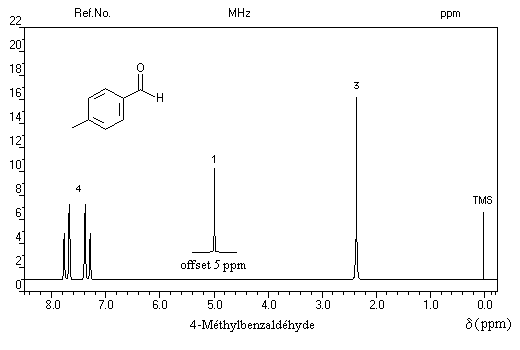
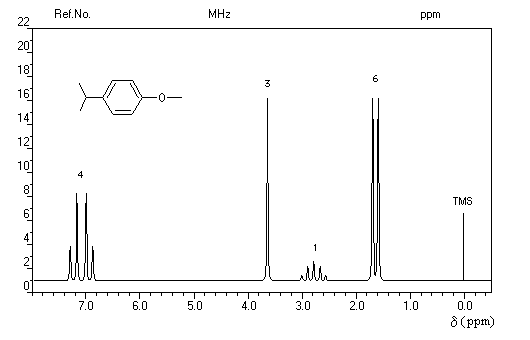
Trois régions sont particulièrement intéressantes :
- 3030 cm-1 : vibrations d'élongation des H du phényle
- 1500 cm-1 - 2000 cm-1 : vibrations d'élongation du squelette aromatique
- 650 cm-1 - 1000-1 : vibrations de déformation hors du plan des liaisons C-H
Composé
|
ortho
|
méta
|
para
|
s (cm-1)
|
730-770
|
750-810
|
800-860
|
Complexes p Les solutions de diiode dans le benzène sont violettes. Mais une étude fine du spectre d'absorption de la solution montre l'existence d'une bande intense dans le proche ultraviolet. Elle est due a la formation d'un complexe p dans lequel le benzène joue le rôle de donneur par son système d'électrons p et le diiode celui d'accepteur.
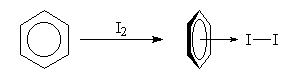

| En dissolvant du diiode dans du benzène on obtient une solution de couleur violette. L'étude du spectre d'absorption montre l'existence d'une bande de transfert de charge : lm = 292 nm ; e = 16 000. Elle est due à la formation d'un complexe p entre le benzène et le diiode. L'expérience de gauche a été réalisée avec du diiode et du toluène. La couleur de la solution est un peu différente de celle obtenue avec le benzène. |
Addition sur le benzéne
Cette réaction a déjà été étudiée dans le cas du benzène à propos de la définition de l'énergie de résonance de la molécule.
Application : hydrogénation sélective des chaînes latérales
Le cycle est thermodynamiquement stable et cinétiquement beaucoup moins réactif qu'une double liaison éthylénique. On peut donc hydrogéner sélectivement une double liaison éthylénique en opérant à température et pression ambiantes. L'exemple suivant concerne le styrène.
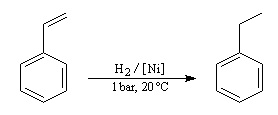
En revanche l'hydrogénation du cycle en présence
d'une double liaison éthylénique n'est pas réalisable par cette voie. On
peut y parvenir indirectement en protégeant la double liaison avant
hydrogénation par synthèse d'un dérivé dibromé. La fonction éthylénique
peut être régénérée après élimination de Br2 par le zinc.
DéshydrogénationLa déshydrogénation d'un cycle est facile lorsque le produit final possède un caractère aromatique. Les catalyseurs de déshydrogénation appartiennent à la même famille que les catalyseurs d'hydrogénation puisqu'un catalyseur catalyse aussi bien la réaction directe que la réaction inverse.
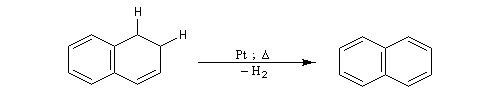
L'addition du dichlore s'effectue en bloc. La réaction est initiée par un rayonnement UV. On obtient différents stéréoisomères de l'hexachloro-1,2,3,4,5,6-cyclohexane. Puisque la molécule possède 6 atomes de carbone asymétriques, on peut prévoir un nombre maximum de 26 = 64 stéréoisomères. Compte-tenu de la symétrie de la molécule il y a seulement 9 stéréoisomères (7 composés méso et un couple d'énantiomères).
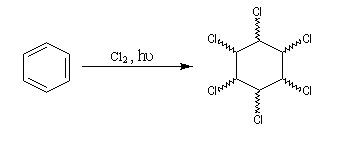
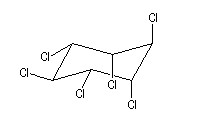 |
L'image de gauche représente le gammexan encore appelé g-lindane en hommage à son découvreur, le chimiste Belge Van der Linden. |
Le cycle central de l'anthracène possède un caractère aromatique plus faible que les deux autres cycles. Il se comporte comme un diène et donne une réaction de Diels-Alder avec des diénophiles suffisamment réactifs comme le tétracyanoéthène TCNE.
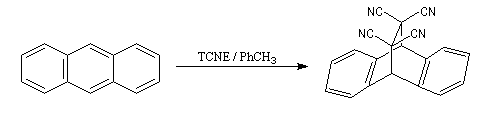
 |
 |
 |
I
|
II
|
III
|
Ces composés sont tous toxiques. L'ensemble est placé sous une hotte ventilée. La réaction se manifeste pendant une période transitoire par une coloration verte due à un complexe de transfert de charge entre l'anthracène (donneur d'électrons) et le TCNE (accepteur d'électrons). Au bout de quinze minutes environ, on obtient le produit qui se présente sous la forme d'un composé solide à la température ordinaire (sublimation à 269 °C). Oxydo-réduction des cycles aromatiques
Oxydation du Benzène L'oxydation du benzène est une réaction d'intérêt essentiellement industriel. On obtient l'anhydride maléique.
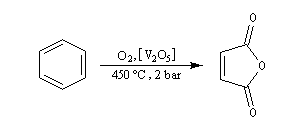
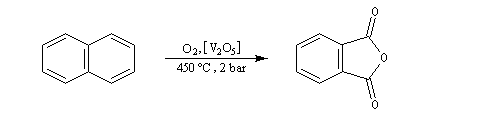
Elle conduit à la formation d'une quinone, l'anthraquinone, ce qui témoigne du faible caractére aromatique du cycle central de cette molécule.
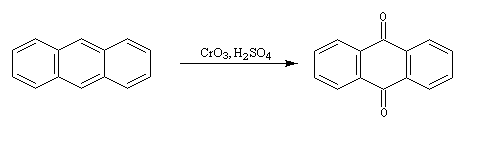
Résultats expérimentaux
La réaction de Birch est une réduction des composés aromatiques par un métal alcalin dissous dans l'ammoniac liquide ou une amine. Il s'agit d'une méthode d'hydrogénation partielle, régiosélective, d'un cycle aromatique [54].
 |
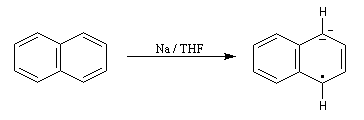
Le naphtalène est un bon accepteur d'électrons vis à vis d'un métal alcalin comme le sodium. Lorsqu'on agite de petits morceaux de sodium avec une solution de naphtalène dans le THF anhydre, on observe la formation d'un anion-radical qui confère à la solution une belle couleur verte. Si l'on ajoute un donneur de protons comme l'éthanol la couleur verte disparaît et il se forme du dihydronaphtalène. La préparation du THF anhydre est décrite dans le chapitre consacré aux éthers. |
- l'un des cycles accepte un électron ce qui conduit à la formation d'un anion-radical responsable de la couleur verte ;
- le radical-anion réagit avec l'éthanol, solvant protogène pour conduire à un radical ;
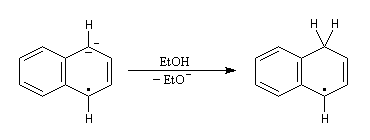
- le radical précédent accepte un nouvel électron pour former un anion ;
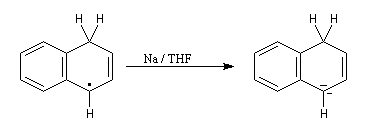
- l'anion réagit à son tour avec le solvant pour donner le produit final.
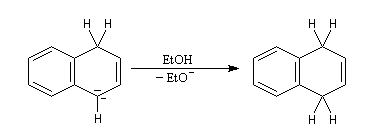
La double réduction de Birch du naphtalène fournit un triène non conjugué bien qu'un composé conjugué soit plus stable. Pourquoi ?
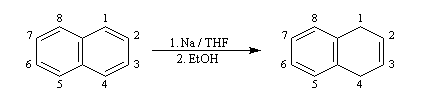
On en déduit les densités électroniques sur les différents sommets des cycles :
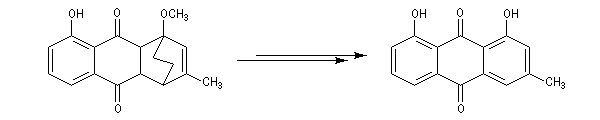
La réaction de Birch est utilisée pour synthétiser des cycles à six chaînons qui sont délicats à obtenir par une autre voie. L'exemple suivant concerne la synthèse de l'acide chrysophanique (de chriso : or et fanein : paraître, en raison de l'aptitude de ce composé à teindre les tissus en jaune), une anthraquinone contenue dans la racine de rhubarbe, selon un protocole mis au point au Research School of Chemistry de l'Australian National University, en Australie, pays d'origine de J. Birch.
-
la réduction partielle du cycle aromatique fournit un diène 1, 4 ;
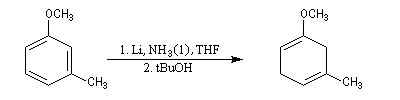
- celui-ci est ensuite isomérisé en diène conjugué plus stable ;
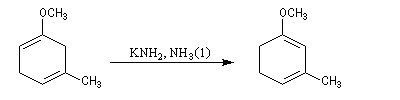
- ce diène sert ensuite de substrat pour une réaction de Diels-Alder.
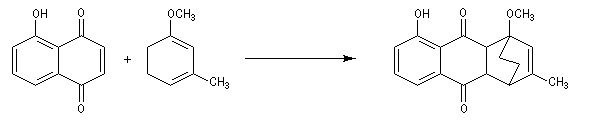
- le reste de la synthèse consiste à aromatiser le troisième cycle et à déprotéger la fonction phénol.
Coupure par oxydation
Réaction
La réaction affecte la chaîne au niveau de l'atome de carbone en a du cycle. Cette position benzilique est très sensible à l'oxydation dans le cas où il y a au moins un atome d'hydrogène. La coupure de la chaine carbonée s'effectue de manière régiosélective quelle que soit sa longueur. L'acide obtenu est donc l'acide benzoïque.
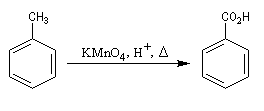
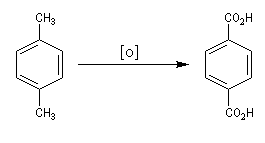
Synthèse de l'acide téréphtalique
L'oxydation du 1,4-diméthylbenzène (p-xylène) par le dioxygène de l'air en présence d'un catalyseur au cobalt, constitue une préparation industrielle de l'acide 1,4-benzènedicarboxylique (acide téréphtalique).
Oxydation de la chaîne latérale du cumène
Le dioxygène s'insère dans la liaison C-H du cumène pour donner un hydroperoxyde de cumyle.
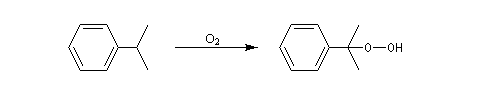
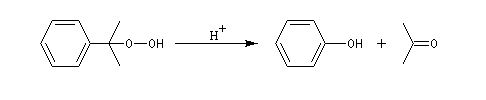
Lorsqu'on porte à reflux du toluène et du dibrome à la température ambiante sous irradiation UV, on obtient facilement le 1-bromo-1-phénylméthane.
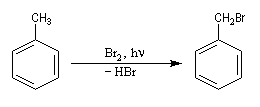
Réactions de substitutions électrophiles
Présentation
Le substrat est le composé aromatique. Le réactif est un électrophile. La réaction consiste en la substitution d'un atome d'hydrogène du substrat par l'électrophile. Il s'agit donc d'une substitution électrophile en série aromatique (rappelons que le caractère électrophile ou nucléophile du mécanisme d'une réaction organique est effectué d'après la nature électrophile ou nucléophile du réactif). Elle est notée SEAr. Puisqu'il s'agit d'une substitution, le caractère aromatique du substrat se retrouve dans le produit final.
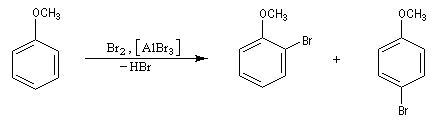
Halogénation
Résultats expérimentaux
- La chloration et la bromation peuvent être effectuées par union directe entre le benzène ou l'hydrocarbure aromatique et les corps simples Cl2 ou Br2 à condition d'utiliser un acide de Lewis : AlCl3, AlBr3, FeBr3 comme catalyseur. Lorsqu'il s'agit de préparer un intermédiaire de synthèse, on préfère généralement la bromation car Br2 présente l'avantage d'être liquide à la température ambiante.
- La fluoration directe est impossible car F2 détruit le substrat par oxydation.
- L'iodation directe est également impossible car I2 n'est pas suffisamment réactif et l'ion iodure formé provoque la réaction inverse. L'iodation est réalisable en utilisant des réactifs qui sont source d'iode cationique comme le chlorure d'iode ICl (réactif de Wijs) ou le mélange I2/ HNO3 (car dans ce cas I- est oxydé par l'acide nitrique). Les dérivés iodés sont obtenus de manière plus commode en utilisant les sels de diazonium comme intermédiaires.
 |
AlCl3 est un composé de couleur blanche, solide à la température ordinaire. Il est très hygroscopique et en présence d'eau ou d'air humide, il est hydrolysé en Al(OH)3 et HCl qui forme un brouillard. Le solide se sublime à 192 °C pour donner une vapeur constituée de molécules de formule Al2Cl6. Cette propriété permet de le purifier. |
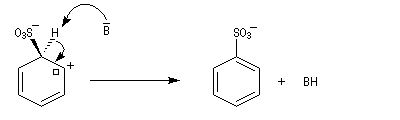
Nous allons raisonner sur l'exemple de la bromation. La réaction s'effectue en plusieurs stades :
- la formation de l'électrophile met en jeu la polarisabilité de la molécule de Br2. L'interaction avec acide de Lewis que constitue le catalyseur, induit une polarisation de la liaison Br-Br ;
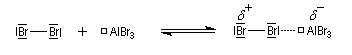
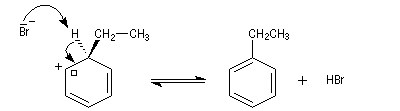
- formation d'un intermédiaire chargé de haute énergie appelé complexe de Wheland ou complexe s. C'est l'étape cinétiquement déterminante de la réaction.
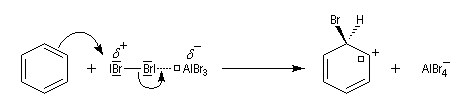
- régénération du catalyseur ;
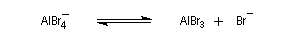
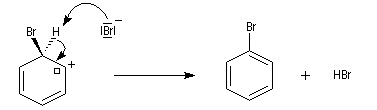
- restauration de l'aromaticité ; Br- joue le rôle de base.
Ce complexe s possède une énergie beaucoup plus élevée que les réactifs. Il s'agit d'un système conjugué non aromatique car le nombre d'électrons ne satisfait pas à la règle de Hückel. Il n'est d'ailleurs pas plan. On peut décrire en première approximation sa structure électronique en utilisant la méthode de la mésomérie.
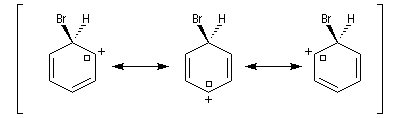
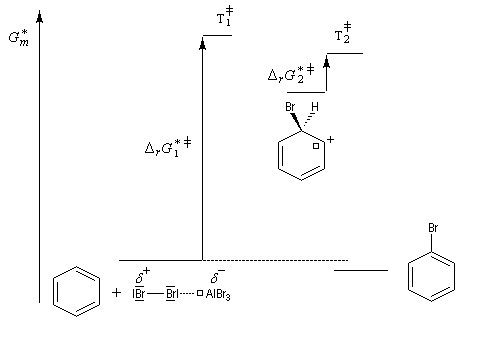
Nitration
Résultats expérimentaux
La réaction de nitration du benzène peut être effectuée à la température ambiante en utilisant HNO3 fumant (titrant 98 % en HNO3) ou le mélange HNO3, H2SO4 concentré. Elle conduit au mononitrobenzène. Ce dernier, plus dense que les réactifs, va au fond du réacteur. Avec des aromatiques plus réactifs comme les phénols, l'acide nitrique dilué suffit mais le mécanisme est différent. Mécanisme
Il s'agit d'une réaction par stades :
- Formation de l'électrophile : l'acide sulfurique joue un
double rôle. C'est un réactif protogène qui améliore le caractère
nucléofuge du groupement -OH de l'acide nitrique en le protonant.
C'est aussi un déshydratant énergique qui permet le déplacement de l'équilibre de formation de l'électrophile en captant l'eau formée.
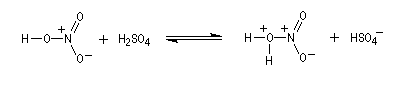 Le véritable réactif électrophile est le cation nitronium NO2+. On dispose de plusieurs preuves de l'existence de cet ion comme agent nitrant. L'une d'elles est le fait qu'on peut utiliser comme réactif de nitration, le mélange : NO2BF4/BF3. Le chimiste américain d'origine hongroise George A. Olah a aussi montré le rôle de l'hexafluoroplatinate de nitronium NO2+PF6- qui apporte directement le cation nitronium NO2+. Avec le benzène, en milieu homogène (solvant CH3NO2), cette étape est cinétiquement déterminante. G. Olah a obtenu le prix Nobel en 1994 [33].
Le véritable réactif électrophile est le cation nitronium NO2+. On dispose de plusieurs preuves de l'existence de cet ion comme agent nitrant. L'une d'elles est le fait qu'on peut utiliser comme réactif de nitration, le mélange : NO2BF4/BF3. Le chimiste américain d'origine hongroise George A. Olah a aussi montré le rôle de l'hexafluoroplatinate de nitronium NO2+PF6- qui apporte directement le cation nitronium NO2+. Avec le benzène, en milieu homogène (solvant CH3NO2), cette étape est cinétiquement déterminante. G. Olah a obtenu le prix Nobel en 1994 [33].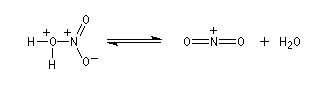
- Deuxième stade : formation de l'intermédiaire de Wheland.
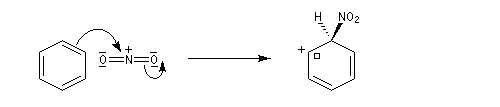
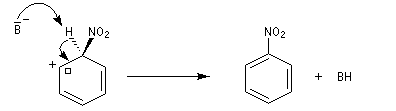
- Troisième stade : restauration de l'aromaticité.
L'absence d'effet isotopique primaire, montre que l'arrachage de
l'atome d'hydrogène par la base n'a pas lieu dans l'étape cinétiquement
déterminante. B- symbolise une base du milieu réactionnel HSO4- ou NO3-.
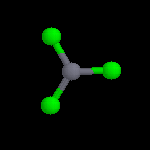
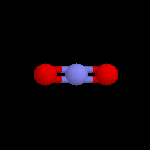
L'image de gauche représente le cation NO2+. Conformément aux prévisions de la
méthode VSEPR, cet ion est linéaire. Sa présence dans les réactifs nitrants a été montrée de plusieurs manières :
|
Les dérivés polynitrés, notamment le 2,4,6-trinitrotoluène, sont utilisés comme explosifs. On les obtient dans des conditions plus dures que les mononitrés. La réaction doit être effectuée à chaud et sous pression.
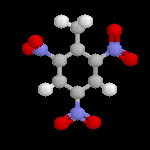 |
L'image de gauche représente la molécule de trinitrotoluène plus connue sous le sigle TNT, encore appelée tolite. A la différence de la nitroglycérine, c'est un composé assez stable pour lequel la décomposition nécessite un explosif auxilliaire. Comme tous les explosifs nitrés, la réaction est une oxydo-réduction interne qui provoque une grande libération de gaz. L'augmentation brutale de volume qui en résulte est à l'origine d'une onde de choc et du caractère brisant de l'explosif. |
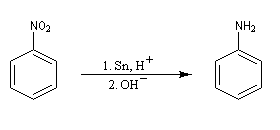
Les dérivés nitrés aromatiques sont une voie d'accès intéressante aux amines aromatiques par réduction du groupe nitro. L'introduction directe d'un groupe amino n'est en effet possible que sur des cycles fortement appauvris en électrons ou en utilisant des bases très fortes, en suivant un mécanisme de substitution nucléophile aromatique SNAr.
La réduction du nitrobenzène par un métal en milieu acide, conduit à un sel d'anilinium. Après passage en milieu basique, on obtient l'aniline.
Les équations redox sont détaillées ci-dessous dans le cas de la transformation suivante.
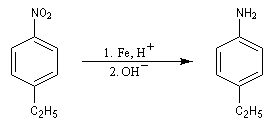
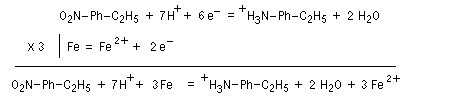
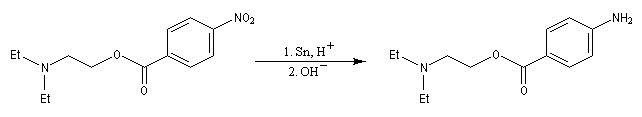
Résultats expérimentaux
La sulfonation du benzène peut être effectuée en portant à reflux un mélange de benzène et d'oléum (SO3 dissous dans H2SO4 concentré).
Mécanisme
Il s'agit d'une réaction par stades. On va se placer dans le cadre d'un contrôle cinétique de la réaction.
- Formation de l'électrophile : l'électrophile est le trioxyde de soufre SO3 ou sa forme protonée HSO3+. Nous allons raisonner avec SO3.
On peut écrire plusieurs formes mésomères, pour SO3 : puisque O est plus électronégatif que S, les formes qui possèdent un atome de soufre déficient en électrons ont un poids statistique élevé.
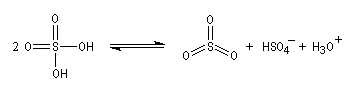
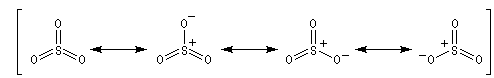
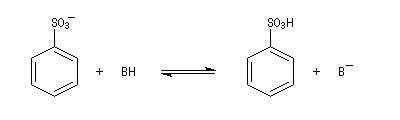
- Formation de l'intermédiaire de Wheland.
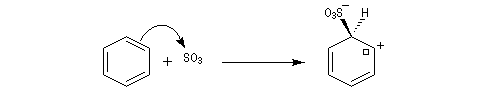
- Réaromatisation.
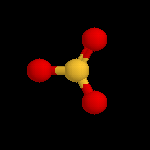 |
L'image de gauche représente la molécule de SO3. A la température ordinaire, le trioxyde de soufre est un liquide qui bout à 45 °C. A l'état solide, ces molécules ont tendance à former des trimères de formule S3O9. L'acide sulfurique concentré à 98 % est capable de dissoudre SO3 pour donner un liquide appelé oléum ou acide sulfurique de Nordhausen. |
La réaction de sulfonation présente l'importante caractéristique de pouvoir être renversée. Si l'on traite un acide sulfonique à chaud par une solution étendue d'acide, on observe le remplacement de HSO3+ par H+. D'un point de vue mécanistique, il s'agit d'un exemple de substitution ipso. La force motrice de la réaction est, dans ce cas, la formation des composés les plus stables. La réaction est sous contrôle thermodynamique.
Sulfonation du naphtalène
Par monosulfonation du naphtalène, on peut former a priori deux isomères : l'acide naphtalène-1-sulfonique et l'acide naphtalène-2-sulfonique nommés respectivement a et b.
Lorsque la réaction est effectuée à 50 °C on obtient de façon majoritaire le composé le plus rapidement formé : c'est l'isomère a qui est le produit cinétique de la réaction.
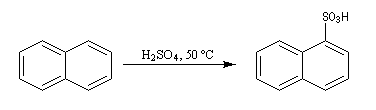
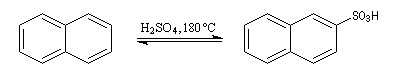
- Substitution en a : deux formes mésomères.
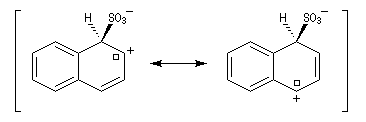
- Substitution en b : une seule forme
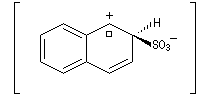
 |
 |
- Le produit P1 (a) se forme le plus vite. L'état de transition qui y conduit est celui qui possède l'enthalpie libre molaire la plus basse. C'est le produit cinétique de la réaction ;
- Le produit P2 (b) est le produit le plus stable. Il lui correspond l'enthalpie libre la plus basse DrG* < 0). C'est le produit thermodynamique de la réaction ;
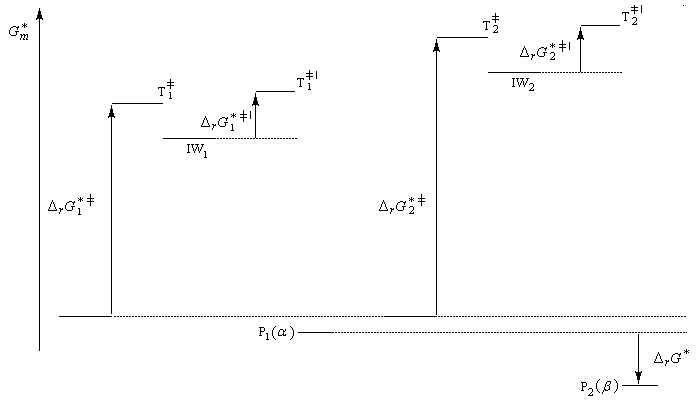
En raison du caractère renversable de la réaction, un acide sulfonique peut être désulfoné par hydrolyse à température élevée. Cette caractéristique permet d'utiliser la suite sulfonation-désulfonation pour bloquer temporairement une position d'un cycle aromatique dans une synthèse multiétapes.
Les acides sulfoniques sont des acides forts qui possèdent de nombreuses applications. L'acide paratoluènesulfonique, en abrégé APTS est utilisé pour préparer des esters sulfoniques d'alcools. Outre leur utilisation en synthèse, les acides benzène-sulfoniques sont utilisés comme détergents. Un des gros avantages des détergents par rapport aux savons est de ne pas précipiter sous forme de sels en eau dure (Ca2+, Mg2+). Notons que la fusion alcaline des dérivés sulfoniques conduit aux phénols dans des conditions assez drastiques.
Alkylation de Friedel et Crafts
Résultats expérimentaux
Il s'agit d'une réaction de création de liaison carbone-carbone. Le réactif alkylant possède un atome de carbone déficient en électrons donc électrophile (dérivé halogéné, alcool, composé éthylénique en milieu acide etc.)
La réaction nécessite la plupart du temps l'emploi d'un acide de Lewis comme catalyseur afin d'exalter le caractère électrophile du réactif. On utilise AlCl3 ou AlBr3 avec les dérivés halogénés. H+ est aussi utillisé avec les alcools et les composés éthyléniques.
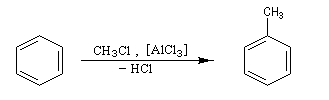
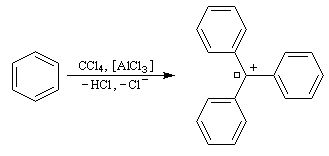
 |
Réaction de Friedel et Crafts entre le toluène et CCl4. Dans un tube à essais bien sec on introduit une spatule de chlorure d'aluminum anhydre. On chauffe le fond du tube jusqu'à ce que AlCl3 se vaporise et se condense sur la paroi froide en haut du tube. Lorsque le tube est revenu à la température ambiante, on verse au moyen d'une pipette Pasteur quelques gouttes d'un mélange de toluène et de tétrachlorométhane CCl4. Une couleur rouge sang se développe au niveau du catalyseur qui témoigne de la formation d'un carbocation fortement conjugé. |
Mécanisme
Il s'agit d'une réaction par stades mais, l'alkylation de Friedel et Crafts est sous contrôle thermodynamique. Les différentes étapes sont renversables. On va raisonner avec un dérivé halogéné.
- Formation de l'électrophile :
La réaction entre le dérivé halogéné et un acide de Lewis (AlCl3 , AlBr3) conduit à un carbocation potentiel (ou à un vrai carbocation dans le cas où ce dernier est suffisamment stable. Par exemple l'écriture CH3+ que l'on rencontre parfois est simplificatrice.)
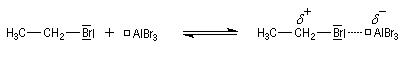
- Formation de l'intermédiaire de Wheland.
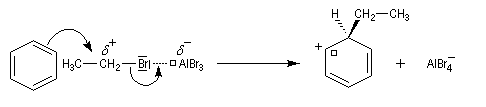
- Réaromatisation.
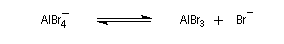
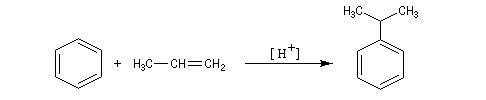
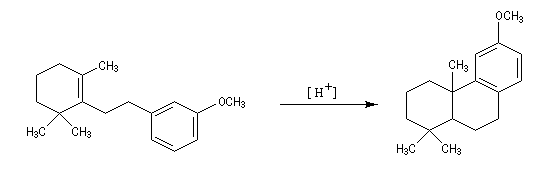
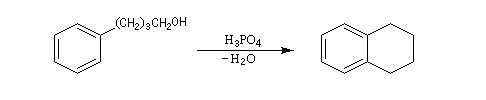

- la tendance marquée des carbocations aux transpositions : primaire ® secondaire ® tertiaire ;
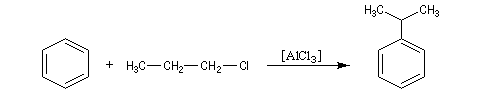
- il est très difficile d'éviter la polyalkylation car les dérivés alkylés réagissent plus rapidement que le benzène lui même ;
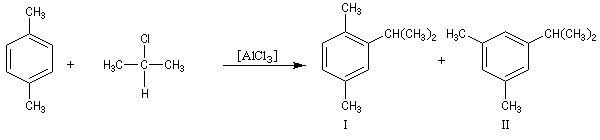
- les différentes étapes du mécanisme étant renversables, la transformation peut faire l'objet d'un contrôle thermodynamique. Dans ce cas, le composé le plus stable est alors majoritaire. Dans l'exemple ci-dessous, il s'agit du composé II.
Résultats expérimentaux
Il s'agit d'une synthèse de cétone aromatique. Le réactif acylant est souvent un halogénure d'acyle. La réaction nécessite l'utilisation d'un acide de Lewis comme AlCl3 en tant que catalyseur.
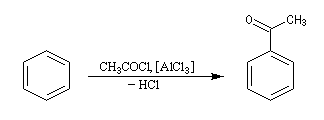
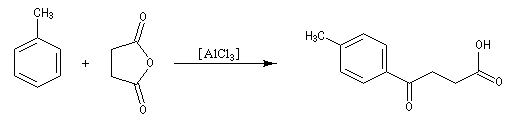
On va raisonner dans le cas où le réactif acylant est un halogénure d'acyle. On distingue trois étapes :
- Formation de l'électrophile : le caractère électrophile
du carbone du chlorure d'acyle est exalté par la formation d'un complexe
avec le catalyseur.
On peut aussi observer l'intervention d'un véritable cation acylium.
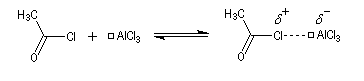
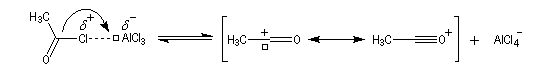
- Formation de l'intermédiaire de Wheland.
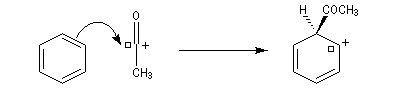
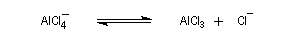
- Restauration de l'aromaticité.
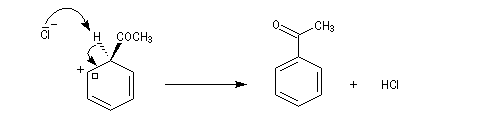
Quand on effectue l'acylation en présence d'un catalyseur acide de Lewis comme AlCl3, il est nécessaire d'utiliser au moins une mol de catalyseur par mole de cétone formée. En effet celle-ci est une base de Lewis suffisamment forte pour former un complexe avec AlCl3 qui bloque son activité catalytique.

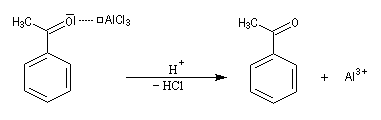
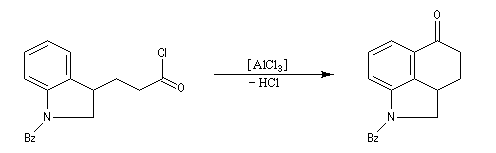
Réaction d'acylation intramoléculaire
Dans les cas favorables (cycles à 5 ou 6 chaînons), les réactions d'acylation peuvent être mises à profit pour réaliser des cyclisations, comme dans l'exemple suivant.
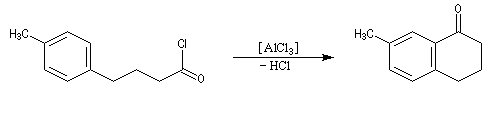
L'acétylation du ferrocène peut être effectuée facilement en présence d'anhydride acétique à chaud. Le ferrocène étant beaucoup plus réactif qu'un aromatique ordinaire, son acylation ne requiert pas la présence d'un acide de Lewis comme AlCl3. L'acide phosphorique concentré suffit.
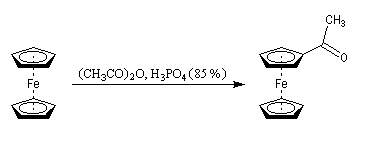
On obtient également une petite quantité de produit diacétylé. Puisque le groupe acyle est désactivant la seconde acylation s'effectue de façon préférentielle sur l'autre cycle.
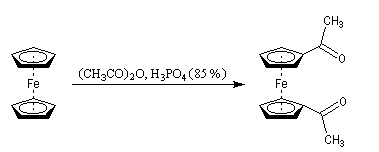
 |
La photographie de gauche représente une plaque chromatographique utilisée pour la mise en évidence des
produits de la réaction d'acylation du ferrocène . L'éluant est un mélange de toluène et d'éthanol (30/1). De gauche à droite :
On trouvera un mode opératoire à la référence [3]. |
Résultats expérimentaux
On peut se demander s'il est possible de synthétiser des aldéhydes en utilisant la réaction de Friedel et Crafts. Le chlorure d'acyle qu'il faudrait utiliser a priori est le chlorure de méthanoyle HCOCl. Ce composé est malheureusement instable. La formylation des hydrocarbures aromatiques est cependant possible en utilisant un mélange de monoxyde de carbone et de chlorure d'hydrogène avec AlCl3 comme catalyseur. La réaction prend alors le nom de réaction de Gattermann-Koch. Avec AlCl3 seul, la réaction est effectuée sous pression et elle est surtout utilisée dans l'industrie. En présence de CuCl, elle est réalisable à la température ordinaire.
Substitutions électrophiles sur des dérivés du benzène Position du problème
On s'intéresse à la réaction de SEAr entre un dérivé monosubstitué du benzène Ph-A et un réactif électrophile. Ce dernier sera noté symboliquement E+ mais il peut s'agir d'une molécule. Nous allons aborder ce problème en l'examinant sous deux aspects :
- aspect cinétique : la réaction peut être plus ou moins rapide que celle mettant en jeu le benzène comme substrat ;
- régiosélectivité : 3 stéréoisomères peuvent être obtenus dans des proportions qui sont a priori différentes.
Position relative
|
1,2
|
1,3
|
1,4
|
Nom
|
ortho
|
méta
|
para
|
Groupe
|
Vitesse relative
|
ortho
|
méta
|
para
|
-OH
|
103
|
40
|
< 2
|
58
|
-CH3
|
25
|
58
|
4
|
38
|
tBu
|
16
|
12
|
8
|
80
|
H
|
1
|
-
|
-
|
-
|
-CH2Cl
|
0,71
|
32
|
15,5
|
52,5
|
-Cl
|
3,3´10-2
|
31
|
< 0,2
|
69
|
-CO2Et
|
3,7´10-3
|
2
|
72
|
4
|
-CF3
|
2,6´10-5
|
6
|
91
|
3
|
-NO2
|
6´10-8
|
5
|
93
|
2
|
-N+(CH3)3
|
1,2´10-8
|
0
|
100
|
0
|
- ceux pour lesquels la vitesse relative est augmentée par rapport au benzène : les groupes sont qualifiés d'activants ;
- ceux pour lesquels la vitesse relative est diminuée par rapport au benzène : ils sont désactivants.
Activants faibles
|
Désactivants faibles
|
Désactivants forts
|
|
-NH2 , -NHR ,
-NR2,
-NHCOR, -OH, -OR
|
alkyle , phényle
|
-F, -Cl , -Br , -I
|
-NO2, -CF3, -N+R3
-CO2H, -CO2R,
-COR -SO3H, -CN |
- les groupes qui orientent en ortho ou para ;
- les groupes qui orientent en méta.
Orientation en ortho-para
|
Orientation en méta
|
activants forts
activants faibles
halogènes
|
désactivants forts
|
Modèle utilisé
Nous allons raisonner dans le cas fréquent où la réaction est sous contrôle cinétique et où la formation du complexe s (intermédiaire de Wheland) constitue l'étape cinétiquement déterminante.
Le postulat de Hammond, indique que l'état de transition de cette étape ressemble à l'intermédiaire, ce qui permet de ramener la discussion de la stabilité de cet état de transition à celle de l'intermédiaire de Wheland. Dans le diagramme ci-dessous, l'intermédiaire IW1 est plus stable que l'intermédiaire IW2. On aura donc : DrG 1* < DrG 2*. Le produit P1 sera plus abondant que le produit P2.
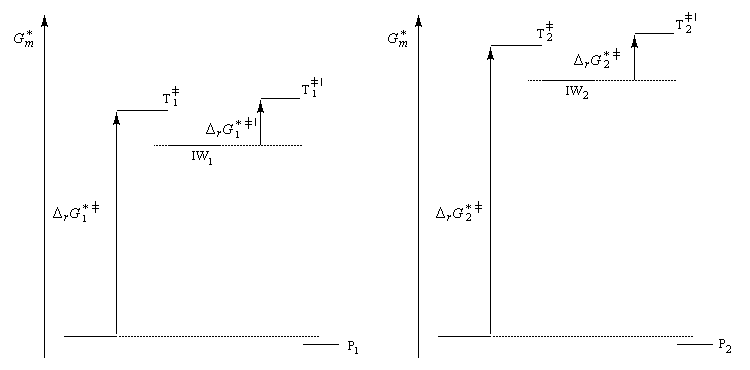
- réactions de sulfonation et d'alkylation de Friedel et Crafts qui sont fréquemment sous contrôle thermodynamique ;
- réactions pour lesquelles l'étape cinétiquement déterminante n'est pas la formation de l'intermédiaire.
Nous allons limiter l'étude à celle des trois cas classiques de substitution sur les positions ortho, méta, para. On connait aussi des exemples de substitutions au niveau de l'atome de carbone qui porte le groupe A. Ce sont des substitutions ipso.
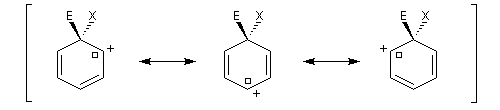
Nous allons comparer la stabilité des différents intermédiaires possibles en utilisant la méthode de la mésomérie.
- substitution ortho
Il apparaît une charge positive au pied du groupe A ;
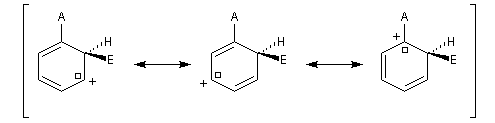
- substitution en para
Il apparaît une charge positive au pied du groupe A ;
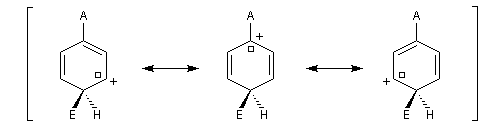
- substitution en méta
Il n'apparaît pas de charge positive au pied du groupe A.
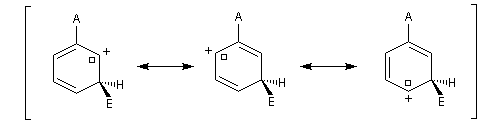
Les groupes interviennent essentiellement par leur effet électronique statique (effet inductif et effet mésomère). On peut distinguer quatre catégories :
Effet électronique de A
|
Cinétique de réaction
|
Régiosélectivité
|
-I & -I, -M
|
désactivants
|
orientation méta
|
- La réaction est ralentie par rapport à ce qu'on observe avec le benzène.
- L'orientation méta est la moins défavorable.
Effet électronique de A
|
Cinétique de réaction
|
Régiosélectivité
|
-I, +M (sauf halogènes)
|
activants
|
orientation ortho-para
|
- la vitesse de la réaction est plus grande qu'avec le benzène ;
- l'orientation ortho-para est favorisée.
Effet électronique de A
|
Cinétique de réaction
|
Régiosélectivité
|
halogènes (-I, +M)
|
désactivants
|
ortho-para
|
Effet électronique de A
|
Cinétique de réaction
|
Régiosélectivité
|
alkyles (+ I)
|
activants
|
ortho-para
|
Le rapport r(ortho)/r(para) dépend beaucoup des conditions expérimentales : par exemple dans la chloration du toluène, ce rapport varie selon les protocoles expérimentaux, de 62/38 à 34/66. Plusieurs facteurs entrent en jeu.
- Le premier est de nature statistique.
Il existe en effet deux positions ortho pour une position para. Sur une base purement statistique on devrait s'attendre à 67 % pour l'isomère ortho et à 34 % pour le para. - Le second est de nature stérique.
On donne ci-dessous les rapports r(o)/r(p) pour la nitration d'alkylbenzènes avec des substrats possédant des chaînes alkyles de plus en plus volumineuses.
Réactionorthoparar(ortho)/r(para)chloration39550,35nitration30700,21bromation11870,06sulfonation< 1> 990,005 - L'isomère ortho peut être favorisé quand une interaction stabilisante intervient entre les deux groupes. Par exemple une liaison hydrogène intramoléculaire, comme dans l'aldéhyde salicylique.
- Quand le groupe orienteur possède un doublet non liant, des effets plus fins peuvent augmenter le pourcentage de para au dépens de l'ortho. Les intermédiaires de Wheland possèdant une structure quinonique para sont en effet plus stables que ceux qui possèdent une structure quinonique ortho. Ce fait est confirmé en RMN qui donne des renseignements sur la distribution des charges sur les différents sommets du cycle.
Il s'agit d'une substitution sur un site déjà porteur d'un groupe autre que l'atome d'hydrogène.
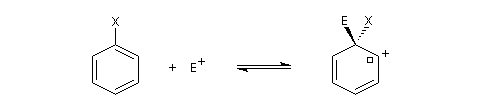
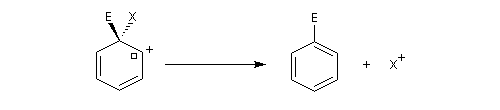
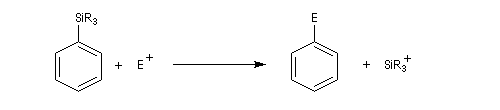
Mécanisme d'addition-élimination
Résultats expérimentaux
Alors que les dérivés halogénés subissent facilement des substitutions nucléophiles en série aliphatique, (mécanismes SN2 et SN1 notamment) les dérivés halogénés aromatiques sont relativement inertes.
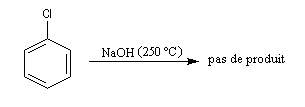
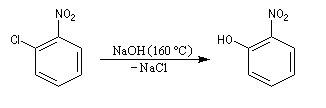
Raisonnons sur le 1-chloro-2-nitrobenzène en tant que substrat.
- Addition du nucléophile et formation d'un complexe (intermédiaire de type Meisenheimer). Cette étape est l'étape
cinétiquement déterminante de la réaction. La relative stabilité de cet intermédiaire est due à la
présence du groupe nitro attracteur (-I, -M).
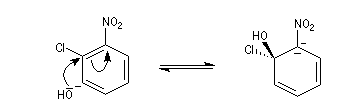
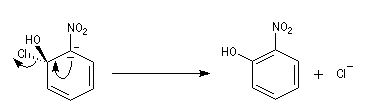
- Elimination du nucléofuge et formation du produit à partir du complexe.
- Synthèse de la 2,4-DNPH
La 2,4-DNPH est un réactif bien connu des composés carbonylés avec lesquels elle forme des 2,4-dinitrophénylhydrazones qui sont généralement facilement cristallisables et dont on peut déterminer le point de fusion. Elle est d'un emploi plus commode que la phénylhydrazine découverte par E. Fischer.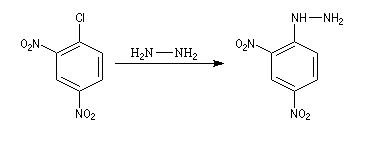
- Identification des amino-acides N-terminaux des protéines
Le dinitrofluorobenzène ou réactif de Sanger (en abrégé DNFB) peut être utilisé pour déterminer la nature de l'amino-acide N-terminal d'une protéine. Le groupe amino est suffisamment nucléophile pour déplacer le fluorure du DNFB par une réaction de SNAr.Après hydrolyse, les fragments sont isolés. L'amino-acide N-terminal est identifié car c'est le seul qui a formé une liaison avec le réactif (les amides ne réagissent pas).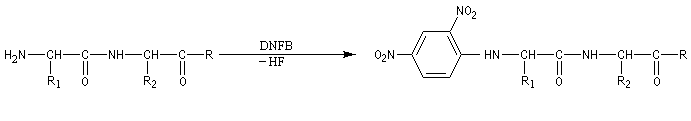
En opérant de la sorte, de proche en proche, on peut remonter à la structure primaire de la molécule. C'est en utilisant une méthode de ce type que le chimiste Britannique F. Sanger (Université de Cambridge) parvint en 1955 à élucider la structure primaire de l'insuline. Ses travaux lui valurent le prix Nobel de chimie en 1958 [34].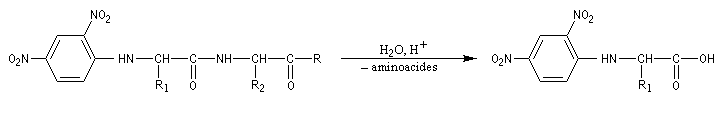
Résultats expérimentaux
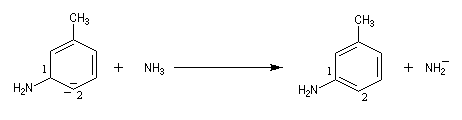
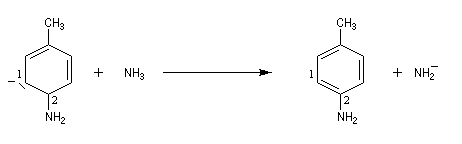
Lorsqu'on fait réagir le parabromotoluène avec l'ion amidure dans l'ammoniac liquide on obtient un mélange de deux amines aromatiques : la paratoluidine et la métatoluidine. La formation de la paratoluidine peut s'expliquer par la substitution d'un atome de brome par un groupe amino. Celle de la métatoluidine est beaucoup plus surprenante car le groupe amino s'est fixé sur une position non occupée par l'halogène sur le cycle aromatique.
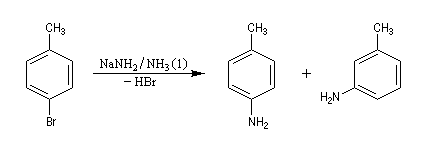
Pour expliquer la formation des produits obtenus, J. D. Roberts (CalTech) a imaginé dans les années 70, l'existence d'un intermédiaire possédant deux points d'attaque : le benzyne. Le mécanisme est le suivant :
- formation de l'intermédiaire benzyne par une réaction d'élimination exigeant une base très forte ;
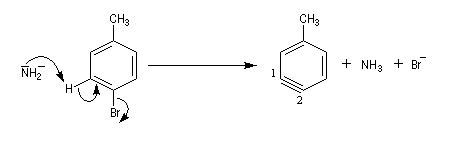
- réaction d'addition du nucléophile sur le benzyne ;
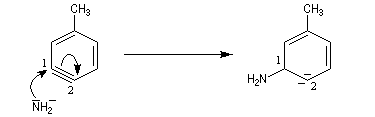
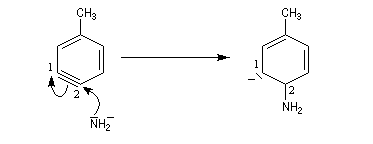
- restauration de l'aromaticité par réaction acide-base ;
| On dispose de nombreuses preuves de l'existence du benzyne comme intermédiaire.
O. L. Chapman et coll l'ont préparé en 1970 par photolyse du peroxyde de phtalyle puis piégé dans une matrice d'argon à 8 K. Le spectre IR montre l'existence d'une bande d'absorption à 1846 cm-1 ce qui témoigne de l'existence d'une liaison dont l'ordre est supérieur à deux et d'évaluer sa longueur : 135 pm. (les distances interatomiques dans le modèle du benzyne représenté à gauche sont approximatives) Zoom : in out Représentation : spacefill ball & stick sticks wireframe |
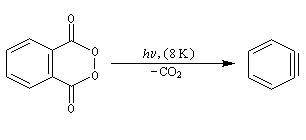
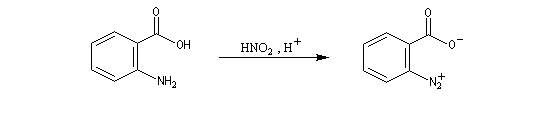
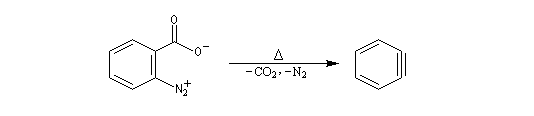
Notons que le benzyne constitue un bon diénophile dans les réactions de Diels-Alder.
Bibliographie
Ouvrages expérimentaux
[1] Manuel d'expériences de chimie - UNESCO Société chimique de France - Université de Montpellier.
[2] P. Rendle, M. V. Vokins, P. Davis, Experimental Chemistry (Edward Arnold).
[3] L. F. Fieser, K. L. Williamson - Organic Experiments (D. C. Heath and Company).
[4] R. Adams, J. R. Johnson, C. F. Wilcox - Laboratory Experiments in Organic Chemistry (The Macmillan Company, Collier-Macmillan Limited).
[5] G. K. Helmkamp, H. W. Johnson Jr, Selected Experiments in Organic Chemistry (W. H. Freeman and Co).
[6] Vogel's Textbook of Practical Organic Chemistry (Longman).
[7] J. R. Mohrig, D. C Neckers, Laboratory Experiments in Organic Chemistry (D. Van Nostrand Company).
[8] R. Q. Brewster, C. A. Van der Werf, W. E. Mc Ewen, Unitized Experiments in Organic Chemistry (D. Van Nostrand Company).
[9] F. G. Mann, B. C. Saunders, Practical Organic Chemistry (Longman).
[10] M. T. Yip, D. R. Dalton, Organic Chemistry in the Laboratory (D. Van Nostrand Company).
[11] Journal of Chemical Education.
Ouvrages théoriques
[12] A. Streitwieser Jr, C. H. Heathcock - Introduction to Organic Chemistry, Macmillan Publishing Co.
[13] J. March - Advanced organic chemistry, Wiley Interscience.
[14] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990.
[15] V. Minkine - Théorie de la structure moléculaire, Editions Mir 1979.
[16] N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
[17] R. Brückner - Mécanismes réactionnels en chimie organique, De Boeck Université 1999.
[18] J. C. Chottard, J. C. Depezay, J. P Leroux - Chimie fondamentale, Hermann 1982.
[19] P. Laszlo - Logique de la synthèse organique. Cours de l'Ecole Polytechnique, Ellipses, 1993.
[20] P. Atkins - Molecular Quantum Mechanics, Oxford University Press 1983.
[21] J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.
Liens
[22] Polycyclic Aromatic Hydrocarbons (PHHs) Data Base in Alphabetic Order
[23] Compléments sur le ferrocène et le cubane
[24] Propriétés chimiques du benzène et des aromatiques
[25] Structure des annulènes
[27] [18]-annulène by F. Sondheimer
[28] Birch reaction
[29] Ferrocene by G. Wilkinson
[30] Ruthenocene by D. E. Bublitz, William E. McEwen, and Jacob Kleinberg.
[31]
[32] G. Wilkinson and E. O. Fischer Nobel prize in Chemistry 1973
[33] G. Olah the Nobel prize in Chemistry 1973
[34] F. Sanger the Nobel prize in Chemistry 1958
[35] Tropylium fluoroborate by Kenneth Conrow
[36] Robert F. Curl, Sir Harold W. Kroto, Richerd E. Smalley The Nobel Prize in Chemistry 1996
[37] Cours G. Février
[38] Cours aromatiques I
[39] Cours aromatiques II
[40] Prismane
[41] The Smalley Institute
[42] Les porphyrines, structure propriétés et applications
[43] Cours sur les aromatiques S. Gerber Ecole Polytechnique de Lausanne.
[44] Introduction à la chimie moléculaire par les orbitales frontières Pascal le Floch, Ecole Poytechnique.
[45] The Royal Institution of Great Britain Site de la Royal Institution.
Articles
[54] A. J. Birch, Quart. Rev. (London), 1950, 4, 69.
[55] T. J. Kealy, P. L. Pauson, Nature, 1951, 168, p. 1039.
[56] F. A. Cotton - l'actualité chimique, juillet 2002 p 29.
[57] Wilkinson G., Rosenblum M., Whiting M. C, Woodward R. B., J. Am. Chem. Soc., 1952, 75, p. 2125.
[58] W. von E. Doering and L. H. Knox, J. Am. Chem. Soc., 79, 352 (1957).
[59] N. H. P. Smith, J. Chem. Educ. 52, 1075, p 238.
[60] Kornfeld, E. A.; Fornefeld, E. J.; Kline, G. B.; Mann, M. J.; Morrison, D. E.; Jones, R. G.; Woodward, R. B. J. Am. Chem. Soc. 1956, 78, 3087.
Aucun commentaire:
Enregistrer un commentaire